Quality Control (QC) instrumentation with common elements is important when needing to show evidence that the final product release for a sterile parenteral batch has met compliance with key shared elements.
Additionally, QC instruments must be compliant with the conditions specified in pharmacopoeias or GMP. Optimization of instruments is also an important factor in ensuring compliance, reducing the risk of human error and preserving the data integrity of test results.
As a guide to ensuring QC compliant batch releases, this article contains an outline of the common QC instrumentation elements, as well as examples of best practice for any instruments used.
Introduction
A compliant final batch release record for sterile parenteral solutions would provide records to prove the following:
- That any water used in the manufacturing of the parenteral was both in control and compliant
- That the manufacturing environment (cleanroom) was in control and compliant
- That regulations regarding contaminating particles in the parenteral itself were adhered to
- That the FDA’s 21CFR part 111 ruling and guidance regarding instrumentation optimization for supporting data integrity in analysis and results was followed
Cleanroom compliance
There are various sources of guidance on good manufacturing practice available that relate to cleanroom compliance when manufacturing sterile parenterals.
For example, the World Health Organisation,2 the European GMP3 and the Pharmaceutical Inspection Co-operation Scheme (PIC/S)4 each state that the air in the cleanroom must be regulated and scrutinized for particles of ≥0.5 microns and ≥5 microns in size.
However, the FDA’s cGMP5 document diverges from this in that it necessitates monitoring of particles ≥0.5 microns.
Regardless of this, any producers of sterile parenteral product need to be aware of the destination marketplace for each batch in production, in order to guarantee that all relevant rules and regulations are being complied with.
Cleanroom monitoring has traditionally been a manual process involving a daily routine of sampling.
Cleanroom monitoring has generally involved manual checks, by moving airborne particle counters around the cleanroom. This means that there is a reliance on the counter operator to demonstrate that the cleanroom was compliant at the time of testing, by ensuring that the test carried out at each location is compliant and correct.
The reliability and accuracy of any final record is also dependant on the operator firstly gathering manual or paper records from that day’s routine environmental monitoring, and then correctly entering these records into the appropriate Excel spreadsheet or secure data repository.
Manual calculations are also required. This is due to the widely-used method of acquiring small samples at each location, and using a relevant factor to multiply these. This then provided the number of particles per cubic meter (m3), as is required by the guidelines. However, the use of such manual processes increases the opportunities for errors to be made.
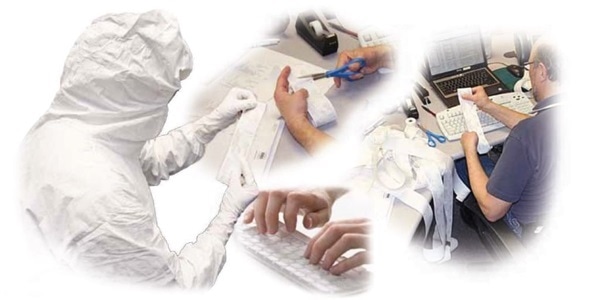
Figure 1. Routine cleanroom environmental monitoring practices are complex and fraught with opportunities for error. Image Credit: Beckman Coulter Life Sciences
Contemporary air particle counters can be optimized for pharmaceutical cleanroom use. In fact, the cleanroom facility’s routine environmental monitoring regime Standard Operating Procedure (SOP) can actually be preconfigured inside the counter.
This removes the reliance on an operator to manually record data such as the sample location name, counter run time, as well as the alarms for each location sampled.
Counters that are optimized for this process can automatically calculate the results per m3. Not only that, but they can also export these results via a secure file transfer, such as File Transfer Protocol (FTP).
This means the data is sent in an electronic format straight to a remote file repository, removing the need for manual data manipulation required by the operator or user. This also provides secure, 21CFR part 11 records to demonstrate that the cleanroom was in compliance during the manufacturing process.
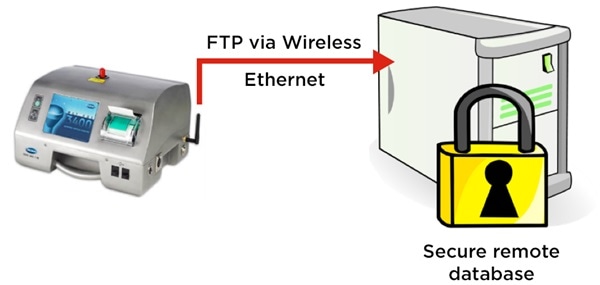
Figure 2. MET ONE 3400 particle counter exports cleanroom routine environmental monitoring electronic records securely via FTP. Image Credit: Beckman Coulter Life Sciences
Final product particulate contamination compliance
According to the United States Pharmacopoeia chapter on therapeutic proteins (USP <787>6), there are three main groups of particles found in parenteral products:
- Intrinsic
- Extrinsic
- Inherent
Contamination via intrinsic particulates generally occurs in the vial or filling process due to inefficient cleaning. Extrinsic particulate contamination tends to occur in the environment where the vial filling occurs.
Contamination via inherent particles is more likely to occur in biopharmaceutical products, where the therapeutic proteins clump together. This can occur through unfavorable environmental conditions, including factors such as bright light, temperatures and extended time periods.
In general, rules around testing for parenteral particulate are harmonized globally, however there can be variations not only from country to country, but also from product to product.
For example, sample volumes for analysis and the format that results are to be reported in can vary from product to product. Sampling requirements for small volume parenteral products like vaccines can differ from large volume parenterals such as an intravenous drip bag.
Results must be calculated and expressed in the correct format, such as counts per container, or counts per mL.
General-purpose liquid particle counting instrumentation can be utilized when testing particles in parenteral products. However, counters that have been optimized for the application are preferable, due to the broad array of complexity in the testing process.
Compendial tests will not only be built into particle counters that have been optimized for this testing, but they can also calculate a pass/fail result automatically.
QC teams generally use their product name to describe the sample under test. Optimized particle counters will allow the user to select the required test for each sample. They do this by allowing the user to select the relevant product by name from a drop-down menu.
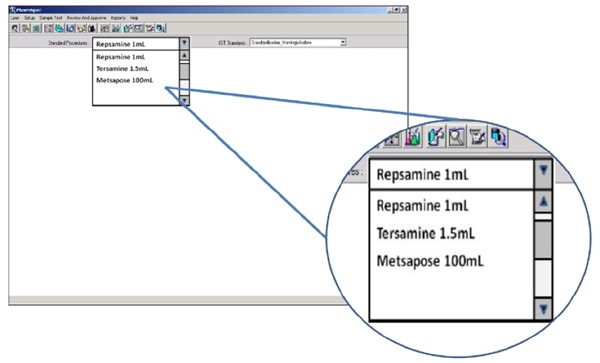
Figure 3. The HIAC 9703+ allows users to select final product quality testing by brand name/product name. Image Credit: Beckman Coulter Life Sciences
While counters that allow operator interface can be located on a local PC, the results database can be automatically stored on a secure remote server. This facilitates access to secure, 21CFR part 11 records needed to show that the batch was in compliance.
Water purity compliance
Water is the main raw material used in the manufacture of parenterals. The relevant quality parameters are not only unambiguously defined in all the major pharmacopoeias, they are in the main harmonized globally.
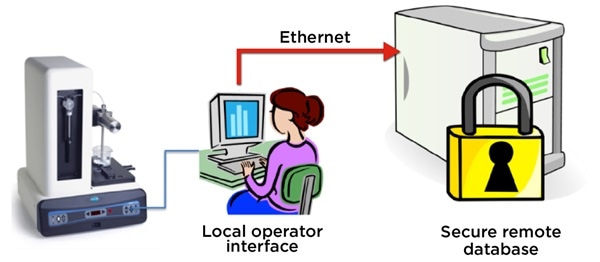
Figure 4. The HIAC 9703+ stores final product quality test result records on a remote, secure server. Image Credit: Beckman Coulter Life Sciences
An important parameter of quality is Total Organic Carbon (TOC). Modern pharmaceutical-grade water systems have will usually have an extremely low TOC content.
This will often be in the low ppb region, in comparison to the amount of Total Inorganic Carbon (TIC) present, usually in the low ppm region, often caused by the increased concentration of dissolved CO2. This is caused through the commonly used water production process known as reverse-osmosis.
General purpose TOC analyzers used to measure TIC and Total Carbon (TC) and calculate the TOC level from these two measurements often struggle to accurately calculate TOC. This is due to the presence of the interfering TIC.
TC ppm (measured) – TIC ppm (measured) = TOC ppb (calculated)
Minor miscalculations in sensor measurements for TC and TIC can lead to large variances in the calculated TOC result. This can cause negative TOC results where the measured and reported TIC value is found to be marginally higher than the reported TC value.
International Conference on Harmonization guideline ICH Q27 contains the relevant outlines for instrumentation validation. The document unequivocally directs the reader to perform an investigation on the specificity of an analytical technique. This is to ensure that it can differentiate between compounds of closely related structures.
Where a user needs to confirm that an analysis method designed to measure TC and TIC and calculate TOC is suitable for the water quality on their site, this guideline can be a useful tool.
In common with other instruments discussed in this article, the capacity to export results via a secure electronic transfer, such as File Transfer Protocol (FTP), to a remote 21CFR part 11 secure data repository is highly advantageous. This is because if helps finalize the optimization requirements for a TOC analyzer used to provide batch release data in the pharmaceutical QC application.
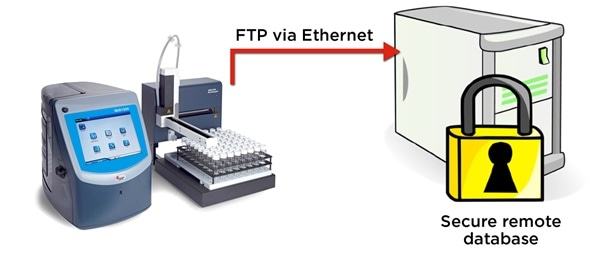
Figure 5. The QbD1200 particle counter exports WFI test records in electronic format securely via FTP. Image Credit: Beckman Coulter Life Sciences
Conclusion
Not only is pharmaceutical QC testing complex, it is also vital for a successful, compliant batch release.
A QC team leader is best advised to look for instrumentation that has been optimized for pharmaceutical QC use. They should take into account factors including automated, pre-configured SOPs, built-in compendial tests and secure electronic transfer, such as File Transfer Protocol (FTP), to ensure 21CFR part 11 electronic record retention.
References
- U.S. Department of Health and Human Services Food and Drug Administration Guidance for Industry, Part 11, Electronic Records; Electronic Signatures — Scope and Application August 2003 U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
- World Health Organisation (WHO) Good Manufacturing Practices For Sterile Pharmaceutical Products, 2009 World Health Organization, CH-1211 Geneva 27, Switzerland.
- European Commission. EudraLex. The Rules Governing Medicinal Products in the European Union. Volume 4. EU Guidelines to Good Manufacturing Practice. Medicinal products for human and veterinary use, Annex 1: Manufacture of Sterile Medicinal Products, 14th February 2008. European Commission Enterprise and Industry Directorate-General, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel – Belgium.
- Pharmaceutical Inspection Co-operation Scheme, PIC/S Guide To Good Practices for The Preparation Of Medicinal Products In Healthcare Establishments, 1st April 2008, PIC/S Secretariat 14, rue du Roveray CH - 1207 Geneva Switzerland.
- Food and Drug Administration. Guidance for industry. Sterile drug products produced by aseptic processing – current good manufacturing practice, 2004. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
- Food and Drug Administration. USP<787> SUBVISIBLE PARTICULATE MATTER IN THERAPEUTIC PROTEIN INJECTIONS, August 1st 2014. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Validation Of Analytical Procedures: Text And Methodology Q2(R1), November 2005 [8th August 2014], http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf [8th August 2014]
- International Society for Pharmaceutical Engineering, The ISPE Good Practice Guide: Ozone Sanitization of Pharmaceutical Water Systems, First edition July 2012 http://www.ispe.org/ispe-good-practice-guides/ozone-sanitization-pharmaceutical-water-systems [14th August 2014]
- Pharmaceutical and Healthcare Sciences Society, Best Practice for Particle Monitoring in Pharmaceutical Facilities, PHSS Technical Monograph No.16, First Edition 2008, ISBN 978-1-905271-15-3
About Beckman Coulter Life Sciences
Beckman Coulter Life Sciences is dedicated to empowering discovery and scientific breakthroughs. The company’s global leadership and world-class service and support delivers sophisticated instrument systems, reagents and services to life science researchers in academic and commercial laboratories, enabling new discoveries in biology-based research and development.
A leader in centrifugation and flow cytometry, Beckman Coulter has long been an innovator in particle characterization and laboratory automation, and its products are used at the forefront of important areas of investigation, including genomics and proteomics.
Primary activity / Product lines
- Flow Cytometry
- Centrifugation
- Particle Counting and Characterization
- Liquid Handling and Robotics
- Nucleic Acid Sample Preparation
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.