Cystic fibrosis is a genetic disorder caused by the dysfunction of a protein that transports sodium and chloride across cell membranes. This protein is called the cystic fibrosis transmembrane conductance regulator (CFTR). Mutation of the CFTR gene gives rise to a protein that lets too much salt and not enough water into the cells, causing mucus to be become thick and sweat to become salty.
A thick, sticky mucus builds up in the respiratory and digestive passageways, which gives rise to symptoms of the condition. Ducts in the pancreas and liver become blocked and the lungs are affected by recurrent infections.
Inheritance of the abnormal CFTR gene
Cystic fibrosis is inherited in an autosomal recessive fashion, which means a person has to inherit two abnormal genes in order for the disease to develop. Everyone inherits two CFTR genes, one from the mother and one from the father. When a person inherits one abnormal copy of the CFTR gene, he or she is a termed a carrier. If that person has a baby with another carrier of a defective CFTR gene, one of the following will apply:
- There is a one-in-four chance that the child will not inherit either of the faulty genes and will have two healthy CFTR genes
- There is one-in-two chance that the child will inherit one faulty gene, making them a carrier
- There is one-in-four chance that the child will inherit both the mutated genes and develop cystic fibrosis
Inheritance Pattern for Cystic Fibrosis
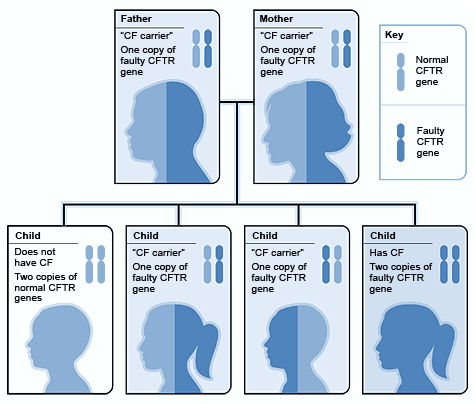
Source: National Heart, Lung, and Blood Institute
Prevalence
In the United Kingdom, 1 person in every 25 carries the faulty gene for cystic fibrosis. In the United States of America, nearly 12 million people have one faulty gene or are carriers for the disease. In the United States, 1 in every 3700 babies born has cystic fibrosis.
The CFTR gene lies on the seventh chromosome and over 1000 different mutations have been discovered. The most common mutation is the ∆F508, which is seen in two thirds of all cystic fibrosis cases worldwide. The mutation is more common among people of northern European descent.
Further Reading
Last Updated: Jan 18, 2023
 New biofilm discovery may pave way to undermining antibiotic-resistant Pseudomonas
New biofilm discovery may pave way to undermining antibiotic-resistant Pseudomonas