Although sickle cell disease (SCD) is present from birth, most patients do not begin to show signs of the condition until after the age of 4 to 6 months. For this reason, many countries use a blood test of newborn babies to screen for the presence of the condition and enable earlier interventions.
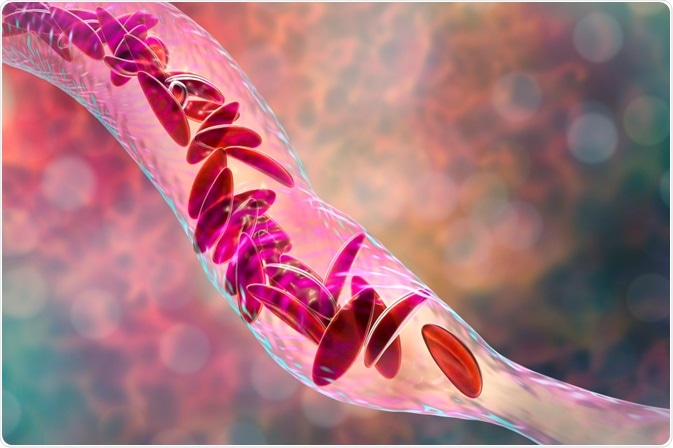
Image Credit: Kateryna Kon / Shutterstock.com
Initial signs
In the beginning, affected patients may experience symptoms such as:
- Dactylitis (swollen, painful hands and feet)
- Fatigue
- Jaundice, particularly yellowing of skin and eyes
The presenting signs are different in each individual, however, according to the severity of the condition and the internal effects.
Sickle crisis
Children with sickle cell anemia do not usually experience serious pain; however, as patients get older, painful episodes called a sickle crisis can develop. A sickle crisis, which can last for a few hours or up to a few days, becomes more common as these patients become older. These crises are characteristic of the condition and involve debilitating pain in various parts of the body such as the lower back, legs, arms, joints, and chest.
Sickle crises are thought to occur due to a disruption in blood flow caused by the rigid hemoglobin in the red blood cells (RBCs). An attack is more likely to occur when the patient has recently been affected by illness, temperature changes, stress, dehydration, or altitude changes. However, the trigger of the crisis is not always clear.
The frequency of episodes can vary greatly among individuals with the condition; some are affected rarely, with a single episode every few years, whereas others have severe crises several times each year.
Anemia symptoms
Sickle cell disease is associated with changes in the hemoglobin and RBCs in the body. In particular, the defected cells have a considerable shorter lifespan as compared to normal RBCs, which can lead to a deficiency in these cells, subsequently causing symptoms of anemia to arise.
Sickle Cell Anemia: A Patient's Journey
These symptoms of anemia can include:
- Fatigue
- Pallor
- Tachycardia
- Dyspnoea
- Jaundice
Blood flow disturbances
In some cases, the rigidity of the defected hemoglobin cells can cause them to become lodged in the blood vessels and disrupt the flow of blood around the body. This can lead to symptoms such as:
- Priapism (prolonged, painful erection)
- Difficulties of vision or blindness
- Transient ischemic attack
- Confusion
- Low leg ulcers
Organ malfunction
Over time, some sickle cell patients may be affected by various symptoms as a result of organs that do not continue to function as required. For example, the spleen stops working properly in some patients, which can lead to a greater susceptibility to infections, including:
- Cholecystitis
- Chlamydia
- Haemophilus influenza type B
- Meningococcus
- Osteomyelitis
- Pneumococcus
- Salmonella
- Staphylococcus
- Urinary tract infection
Other symptoms
As a result of the wide effects of defected RBCs, there are various other symptoms that may occur in certain patients, which may include:
- Delayed growth
- Delayed puberty
- Heart failure due to iron overload
- Abdominal pain, particularly in younger children
- Acute chest syndrome
- Depression
- Joint pain and movement restriction
- Intrahepatic cholestasis
- Gallstones
- Pulmonary hypertension
- Heart disease
There are many complication of sickle cell disease that patients are more likely to experience as a result of the condition. Some of these have a fatal potential and account for the lower life expectancy in these patients as compared to other populations, which is currently between 40 to 60 years in most developed countries.
However, the treatments to manage symptoms and prolong the lifespan of patients with sickle cell disease are continually improving and have already helped to improve the quality of life and symptoms experienced by those with the disease.
References
Further Reading
Last Updated: Nov 28, 2022
 Young adults with complex childhood conditions experience longer hospital stays, higher readmissions
Young adults with complex childhood conditions experience longer hospital stays, higher readmissions