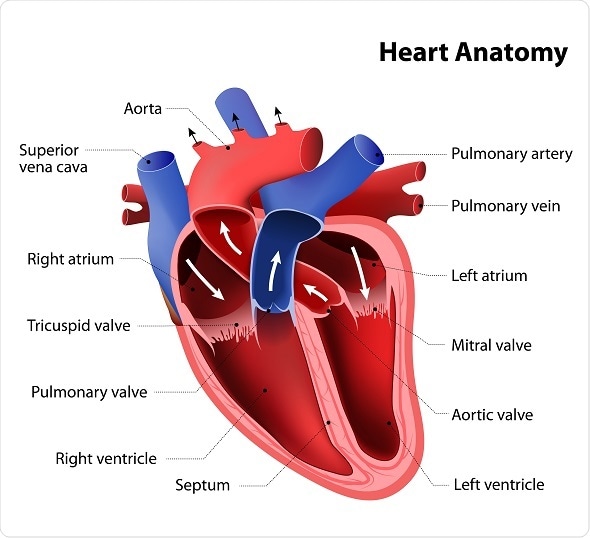
Double outlet right ventricle (DORV) is a rare congenital disorder where both of the great arteries, namely the aorta and the pulmonary artery, arise from the right ventricle. Normally, the aorta arises from the left ventricle where it receives oxygen-rich blood that is sent to the rest of the body. In contrast, the pulmonary artery normally exits from the right ventricle and carries oxygen-poor blood from the heart to the lungs for oxygenation.
©Designua / Shutterstock.com
In addition to DORV, affected infants usually tend to have a hole in the muscular wall that separates the left ventricle from the right. This condition is referred to as a ventricular septal defect (VSD). The location of the VSD in relation to the aorta and/ or pulmonary artery is used to classify the four different types of DORV. Other defects that may also be seen in these infants are pulmonary artery stenosis and transposition of the great vessels.
Clinical presentation
Most infants with DORV develop symptoms within the first early few weeks of life. The clinical picture is similar to what is observed with other types of congenital heart diseases and defects. Parents may note infants having problems with breathing, such as shortness of breath or rapid breathing. Moreover, infants tend to feed poorly and show a general failure with regards to their ability to thrive.
The infant may present with cyanosis, which is a bluish discoloration of the skin that arises due to low oxygen saturation in the blood. Infants may also have other signs and symptoms, such as heart murmurs (i.e., unusual heart sounds), sweating, and fatigue.
The lack of or severity of the pulmonary stenosis is a key determinant to the spectrum of symptoms that the patient will experience. In addition to this, it also determines the age that the patient will present with the clinical picture of DORV. Generally, the neonatal period is the time that most patients present with DORV.
Those with severe narrowing of the pulmonary vessel will have cyanosis, while those with pulmonary blood flow that is poorly controlled will develop congestive heart failure.
Etiology
To date, no predictive factor or specific causal agent has been identified in the development and pathogenesis of DORV. However, like other conotruncal defects of the heart, it is speculated that DORV may have a neural crest origin. The neural crest plays an imperative role in the development of the cardiac septum.
There have been studies demonstrating that completely removing the neural crest during embryonic development results in a truncus arteriosus anomaly. Deleterious effects such as DORV, Eisenmenger complex and tetralogy of Fallot are seen with deletions to smaller parts of the neural crest.
Congenital heart defects are often associated with defects to the thyroid and parathyroid glands as well as the thymus, because these glands are also formed by the same area of neural crest cells responsible for the formation of cardiac structures.
Band 22q11 deletion is seen in tetralogy of Fallot, VSDs, and anomalies of the aortic arch. This deletion, however, is rare in DORV, being seen in only about 1 in every 20 patients affected by the condition. Nonetheless, DORV patients may still be tested for 22q11 deletion, because DORV encompasses a large spectrum of abnormalities.
Further Reading
Last Updated: Feb 26, 2019