Antiviral agents against SARS-CoV-2 are urgently required to combat the ongoing CoV disease 2019 (COVID-19) pandemic. The MPro, also referred to as 3-chymotrypsin-like protease (3CLPro), is a well-known SARS-CoV-2 enzyme. An approved medication, Paxlovid, and various experimental novel drugs, such as S-217622, target the SARS-CoV-2 Mpro. Nevertheless, there is still a need for novel chemical scaffolds targeting MPro, given the inherent drawbacks of the current novel medications and the threat of viral resistance.
About the study
In the present investigation, the researchers focused on the SARS-CoV-2 MPro structure for massive library docking in quest of novel starting points for non-covalent or covalent inhibitor discovery. They virtually coupled 1.2 billion non-covalent and a novel collection of 6.5 million electrophilic compounds against the Mpro enzyme structure from Enamine readily accessible (REAL) space.
The team simulated an aggregate of SARS-CoV-2 MPro coupled to a non-covalent SARS-CoV MPro inhibitor to characterize hot spots for ligand binding across the active site. Three of the refined non-covalent inhibitors' crystal structures, with resolutions spanning from 2.12Å to 2.59 Å, were established to study how the docked positions of the novel inhibitors matched up with their actual binding modes and to guide further refinement.
The authors conducted a second docking experiment because other researchers had discovered efficient inhibitors with comprehensive structure-activity-relationship (SAR) information and scaffolds similar to the ones used in the current work during the research period. They attempted to incorporate the conclusions drawn from the present findings and those of other investigations highlighting the identification of new chemotypes. The COVID-19 Moonshot consortium's non-covalent ligand MAT-POS-b3e365b9-1 (MPro-x11612.pdb), in conjunction with the SARS-CoV-2 MPro crystal structure, was the target of the novel docking screen.
The scientists searched the 1.4 billion compounds in the ZINC15/ZINC20 libraries for three Cys-reactive covalent warheads, nitriles, aldehydes, and alpha-ketoamides to find electrophiles that may covalently alter the catalytic Cys145. DOCKovalent was used to create dockable three-dimensional (3D) molecules for covalent binding.
The crystal structures of five aldehyde inhibitors bound with MPro were developed to examine how the docked positions of the covalent inhibitors correlated with actual binding modes and to support further optimization. While non-covalent and covalent inhibitor optimization progressed, the team evaluated various compounds in a reverse transcription-quantitative polymerase chain reaction (RT-qPCR) viral infectivity test in Henrietta Lacks-angiotensin converting enzyme 2 (HeLa-ACE2) cells.
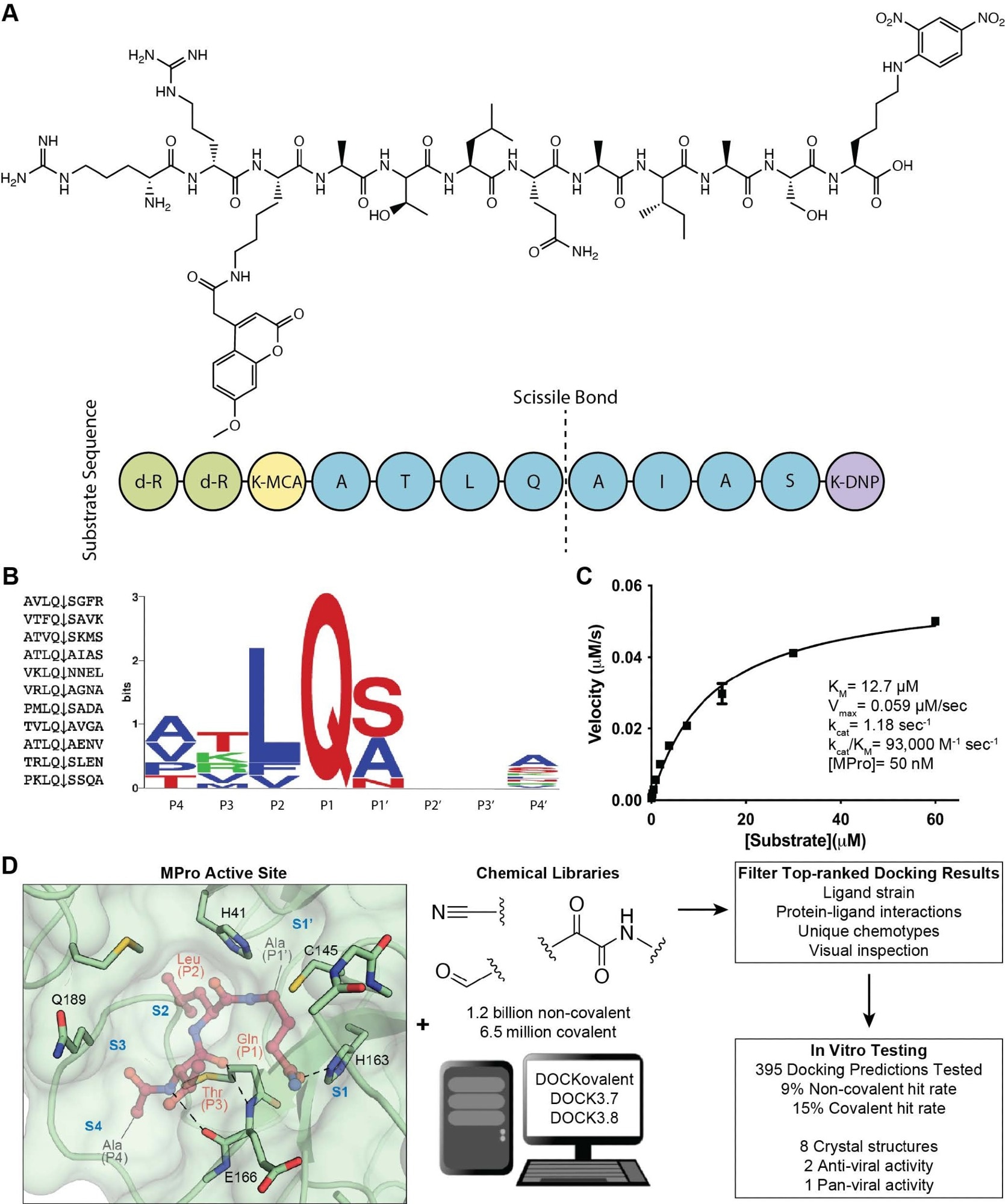 Substrate design and assay development allows structure-based inhibitor discovery. (A) The chemical structure of the optimized NSP7 substrate shown as a schematic (top) of the substrate sequence highlights the role of each residue (bottom). The substrate contains the P4-P4′ NSP7 extended substrate sequence (blue), the fluorophore (yellow), the fluorescent quencher (purple), and the residues for increasing solubility (green). (B) A list of the viral polypeptide NSP sequences (P4-P4′) that are cleaved by MPro (left). The sequenceLOGO highlighting the substrate specificity of MPro, yielding a P4-P4′ consensus sequence: ATLQ(S/A)XXA (right). (C) The Michaelis-Menten kinetics for the NSP7 substrate with MPro yield parameters indicative of an optimized, efficient substrate. (D) SARS-CoV-2 MPro active site (PDB 6Y2G)26 (green; sub-pockets S1′, S1, S2, S3, S4), shown here with substrate preferences (pink; P1′, P1, P2, P3, P4) (modeled after PDB 3SNE)27, was used to dock 1.2 billion non-covalent molecules and 6.5 million electrophile molecules. Top-ranked molecules were filtered and 395 were synthesized for in vitro testing. Some docking hits were prioritized for compound optimization, crystallography, pan-viral enzymatic activity, and cell-based antiviral activity. For C, experiments were performed in triplicate.
Substrate design and assay development allows structure-based inhibitor discovery. (A) The chemical structure of the optimized NSP7 substrate shown as a schematic (top) of the substrate sequence highlights the role of each residue (bottom). The substrate contains the P4-P4′ NSP7 extended substrate sequence (blue), the fluorophore (yellow), the fluorescent quencher (purple), and the residues for increasing solubility (green). (B) A list of the viral polypeptide NSP sequences (P4-P4′) that are cleaved by MPro (left). The sequenceLOGO highlighting the substrate specificity of MPro, yielding a P4-P4′ consensus sequence: ATLQ(S/A)XXA (right). (C) The Michaelis-Menten kinetics for the NSP7 substrate with MPro yield parameters indicative of an optimized, efficient substrate. (D) SARS-CoV-2 MPro active site (PDB 6Y2G)26 (green; sub-pockets S1′, S1, S2, S3, S4), shown here with substrate preferences (pink; P1′, P1, P2, P3, P4) (modeled after PDB 3SNE)27, was used to dock 1.2 billion non-covalent molecules and 6.5 million electrophile molecules. Top-ranked molecules were filtered and 395 were synthesized for in vitro testing. Some docking hits were prioritized for compound optimization, crystallography, pan-viral enzymatic activity, and cell-based antiviral activity. For C, experiments were performed in triplicate.
Results
In the current work, the authors identified 132 SARS-CoV-2 MPro inhibitors belonging to 37 different scaffold classes and exhibit half-maximal inhibitory concentrations (IC50) less than 150 µM. Among these, 15 inhibitors across three scaffolds had IC50 values under 10 µM and inhibited the enzyme. The most effective covalent inhibitor, '7021, was found to function reversibly, which was probably a reflection of the fast-on/fast-off kinetics of aldehyde covalent inhibitors.
Further, the team provided an improved MPro substrate for upcoming inhibitor evaluation. The scientists initially established an electrophile library using the expanding collection of tangible compounds, which included aldehydes, nitriles, and alpha-ketoamides. The community can openly access this library of more than 6.5 million new electrophiles at https://covalent2022.docking.org.
Eight of the novel inhibitors' crystal structures closely matched the docking estimates. The antiviral activities of two of the novel aldehyde inhibitors were comparable to their enzymatic IC50 values, indicating that further optimizing this class for on-enzyme efficacy may predict antiviral activity.
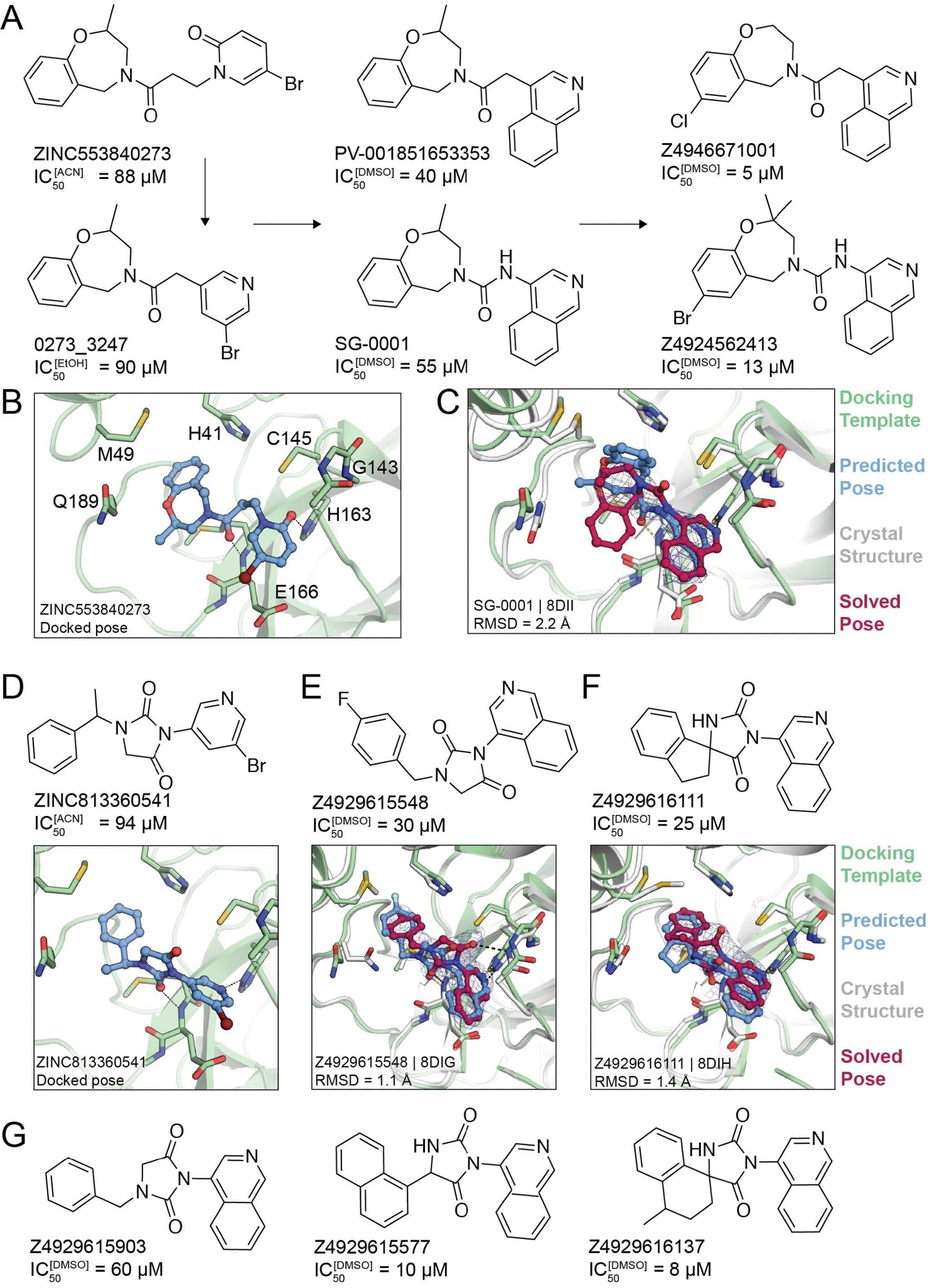 Non-covalent compound optimization to low-μM potencies. (A) Progression of the ‘0273 scaffold. (B) Predicted binding pose of ‘0273. (C) Comparison of crystal structure (grey protein, red compound) and docked complex (green protein, blue compound) of SG-0001 (PDB 8DII). (D) Predicted binding pose of ‘0541. (E), (F) Comparison of crystal structures and docked complexes of ‘5548 (PDB 8DIG) and ‘6111 (PDB 8DIH), respectively. (G) Additional ‘0541 analogs with improved affinities. The 2fo-fc ligand density maps (blue contour) are shown at 1 s. Hungarian root mean square deviations (RMSD) were calculated with DOCK6.
Non-covalent compound optimization to low-μM potencies. (A) Progression of the ‘0273 scaffold. (B) Predicted binding pose of ‘0273. (C) Comparison of crystal structure (grey protein, red compound) and docked complex (green protein, blue compound) of SG-0001 (PDB 8DII). (D) Predicted binding pose of ‘0541. (E), (F) Comparison of crystal structures and docked complexes of ‘5548 (PDB 8DIG) and ‘6111 (PDB 8DIH), respectively. (G) Additional ‘0541 analogs with improved affinities. The 2fo-fc ligand density maps (blue contour) are shown at 1 s. Hungarian root mean square deviations (RMSD) were calculated with DOCK6.
Conclusions
Overall, the team found 37 different scaffolds of SARS-CoV-2 Mpro inhibitors, with the most effective covalent and non-covalent compounds having an IC50 of 20 and 29 μM, respectively. Inhibitors with low micromolar range activity were established after the optimization of several series. The docking projected binding modes were validated later by crystallography, which may serve as a template for future optimization. The compounds identified in the present work reveal novel chemotypes that assist in the future searching for MPro inhibitors for SARS-CoV-2 and other potential CoVs.
 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference:
- Preliminary scientific report.
Large library docking for novel SARS-CoV-2 main protease non-covalent and covalent inhibitors; Elissa A Fink, Conner Bardine, Stefan Gahbauer, Isha Singh, Kris White, Shuo Gu, Xiaobo Wan, Beatrice Ary, Isabelle Glenn, Joseph O'Connell, Henry O'Donnell, Pavla Fajtova, Jiankun Lyu, Seth Vigneron, Nicholas J Young, Ivan S Kondratov, Anthony J O'Donoghue, Yurii Moroz, Jack Taunton, Adam R Renslo, John J Irwin, Adolfo Garcia-Sastre, Brian K Shoichet, Charles S Craik. bioRxiv preprint 2022, DOI: https://doi.org/10.1101/2022.07.05.498881, https://www.biorxiv.org/content/10.1101/2022.07.05.498881v1
 Can an allergy spray help prevent COVID-19?
Can an allergy spray help prevent COVID-19?