Sponsored Content by PittconJan 30 2019
An interview with Dr. Michael Dong, about his upcoming talk at Pittcon.
Why are stability-indicating methods important in pharmaceutical analysis?
The stability-indicating assay is perhaps the single application of HPLC that has the most impact. It is the method used to establish shelf-life for pharmaceuticals. It is this HPLC assay that determines the expiry date of a drug. A pharmaceutical company will conduct thousands of stability studies to establish the shelf lives of their products.

© Zolnierek/Shutterstock.com
HPLC and mass spectrometry each account for only about $5 billion of global sales. In contrast, pharmaceutical global sales are about $1.3 trillion. Even though the value of HPLC equipment sales is minuscule compared to pharmaceutical sales, HPLC is very important in affecting health and well-being when it comes to the drug development process and production.
Before a drug can be marketed it needs to have proven safety and efficacy - both depend on the purity of the drug. Impurities can make the drug unsafe and the level of drug potency depends on how much of the active pharmaceutical ingredient (API) is present. In order for pharmaceutical companies to be certain of the purity of their drugs they conduct numerous quality control analyses.
Acceptable potency is specified for each drug using the HPLC stability-indicating assay, which ensures that each batch of drug product is within 5-10% of the target level (label claim). Similarly, it must confirm that the level of impurities is acceptably low, for example, a maximum of 0.1 - 1% of the total drug. HPLC is a primary tool for the quality control of pharmaceuticals, for both large and small molecules.
The stability-indicating assay for drug purity is important because the purity of drugs is equated with quality and safety. Stability studies are required by regulatory bodies such as the US FDA (United States Food and Drug Administration), or the EMA (European Medicines Agency) in Europe. The purity of the drug must be within specification before it can be sold (release testing) and within the stated expiration date under the recommended storage conditions. Consequently, HPLC is pivotal to the pharmaceutical industry and hundreds of thousands of HPLCs are being used for this purpose alone. More than 50% of HPLC is in the pharmaceutical industry.
Why is RPC/UV a commonly used stability-indicating method and what are the challenges associated with it?
When liquid chromatography was first implemented, a silica column was used. The sample was diluted in a solvent, such as hexane, and poured through a column of silica. This is referred to as normal-phase chromatography. Differential adsorption of components of the sample to the silica resulted in different compounds move through the column at different rates, allowing them to be separated into fractions.
In order to avoid contamination of the column when compounds with particularly strong absorption to silica are present, reverse-phase chromatography was developed. This involves modifying the silica surface to make it hydrophobic and diluting the sample in aqueous solution. It is the reversed of normal phase as the mobile phase is polar and the stationary phase is nonpolar.
Reverse-phase chromatography is based on hydrophobic interaction, which is a much weaker interaction through van de Waal or dispersive forces. This provides reassurance that everything that goes into the column comes out again; there is 100% mass balance. This is critical for drug quality control that no impurities are retained or absorbed in the column. That is one reason that stability-indicating and purity assays invariably use reverse-phase chromatography in addition to the fact that it is highly reproducible and versatile.
Regulations stipulate that the HPLC method must be able to separate the main component, the API, and all impurities including degradation products. All the small molecule pharmaceutical drugs are made by a synthetic process to obtain a highly-purified compound. However, there will always be some trace impurities and each impurity will form a distinct band or peak on the chromatogram. The area under the different peaks in the HPLC chromatograms are proportional to the concentration of the trace impurities.
Pharmaceuticals, even though they are highly stable, do degrade forming degradation products. These too are measured using the stability-indicating method. The chromatography method may need to be tweaked many times in order to achieve a separate peak or zone from the API, the excipients, each impurity and the degradation product, which is essential to accurately quantify each of these components. Good sensitivity, down to at least 0.05% is also needed.
Once the HPLC method has been developed, it must be reproducible for many laboratories around the world as most companies do global manufacturing. For example, in a stability study, a drug may be in the chamber for two to three years, and needs to be measured every three months, year after year. The bar for method performance, accuracy, and reproducibility, is thus very high. So, not only is it HPLC challenging, it is also under very stringent regulatory compliance guidance.
The challenges for HPLC assay are increasing as newer drugs get more complex. More than 50% of newly developed pharmaceuticals are chiral-compounds with many having multiple chiral centers. Consequently, it is not unusual to have 40-60 different peaks in the HPLC impurity profiles of a complex drug. These more complex drugs require greater separation power in the HPLC column. Development of such methods are time-consuming and arduous, and the analysis time keeps getting longer.
What is ultra-high-pressure liquid chromatography (UHPLC) and how is it beneficial over conventional HPLC?
Conventional liquid chromatography uses gravity to draw the diluted sample through the packed column, so the different fractions can be collected. It's a purification technique and is not a very reproducible process.
High performance liquid chromatography uses a column with smaller particles and the whole process is automated using a high-pressure pump. For about 50 years, the pressure limit was 6,000 psi (400 bars). As more complex separations were needed, for example to analyse complex drugs, environmental samples, physiological fluid bloods and serum, or in proteomic studies, there was a need for greater separation power and faster analysis.
In 1997 Professor Jim Jorgenson in University in North Carolina at Chapel Hill demonstrated that liquid chromatography can be performed at ultra-high pressures to provide higher speed and resolution. Pressure, and separation power, is controlled by the particle size in a column, and by this time particle size had been reduced to below 2 microns.
In 2004 the first commercial UHPLC instrument was developed by Waters Corporation, who you will be able to visit in the exhibition floor at Pittcon 2019. HPLC conducted at pressures above 15,000 psi is generally referred to as ultra-high-pressure liquid chromatography (UHPLC).
HPLC can separate a maximum of about 200 peaks, whereas UHPLC can separate almost 1,000. This makes it possible to quantitate very complex drugs much more confidently and accurately.
There are also other features of UHPLC that make it better. Typically, they have a smaller flow path (system dispersion). The UV detector flow cell is smaller; less than one microliter, compared with 8-10 microliters in HPLC. A smaller flow cell controls dispersion or band broadening so that narrow peaks are maintained.
What is the 3-pronged template approach to HPLC method development and why is it well suited to early drug development?
This is a terminology I have coined. When I was working for a major pharmaceutical company, many scientists were developing HPLC methods to support his or her new drug development projects and everybody did it their own way. A more efficient process was needed for the development of high-quality methods with more consistency for the less experienced scientists. My proposed solution was this three-pronged template approach. If you have a template you can follow, the method development process is a little easier and encourages consistency across individuals with different experience levels. This would be especially helpful for the stability-indicating assay using template #3 (multi-segment gradient approach).
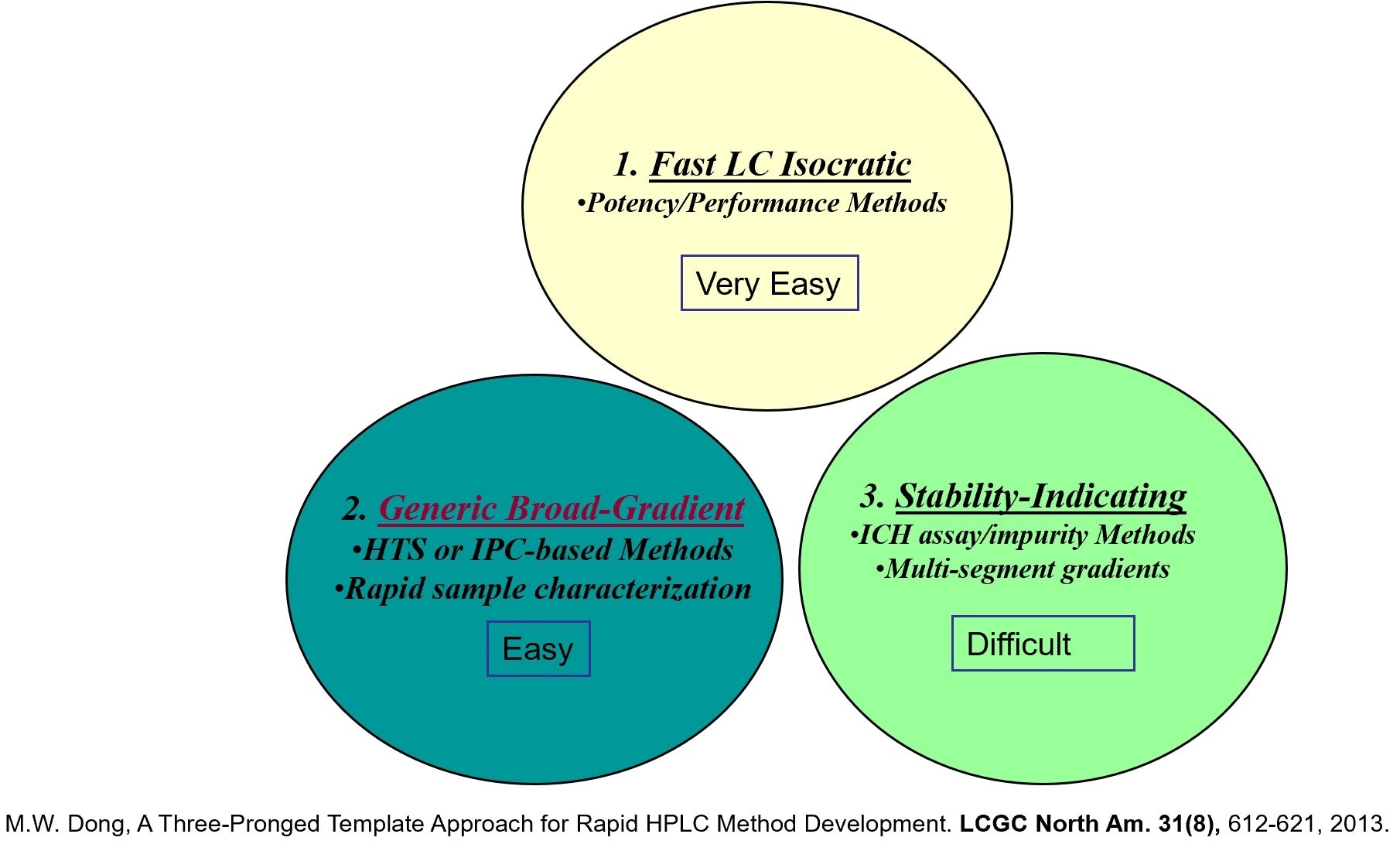
These three templates help laboratory scientists to understand the different method types and decide which method template is appropriate for the intended use. It provides a model to help reach the end result faster by understanding the method attributes for each template.
Please describe your research which led you to develop this concept.
My research was much more applied research than basic research. Working for the pharmaceutical industry, I developed many HPLC methods in rapid successions.
At that time, I was supporting an oncology project for a drug for prostate cancer and gastric cancer. The molecule is quite complex and has 3 chiral canters, and many methods must be developed quickly to support the synthetic process to control the purities of the starting materials, intermediates and the API. I applied the three-pronged template approach in this project and developed 40+ methods to support this very complex project efficiently.
The use of UHPLC method development is a no-brainer; UHPLC is faster, because it uses shorter columns packed with smaller particles. I am therefore going to talk more about the #3 template approach of using a multi-segment gradient and how to apply it in two case studies.
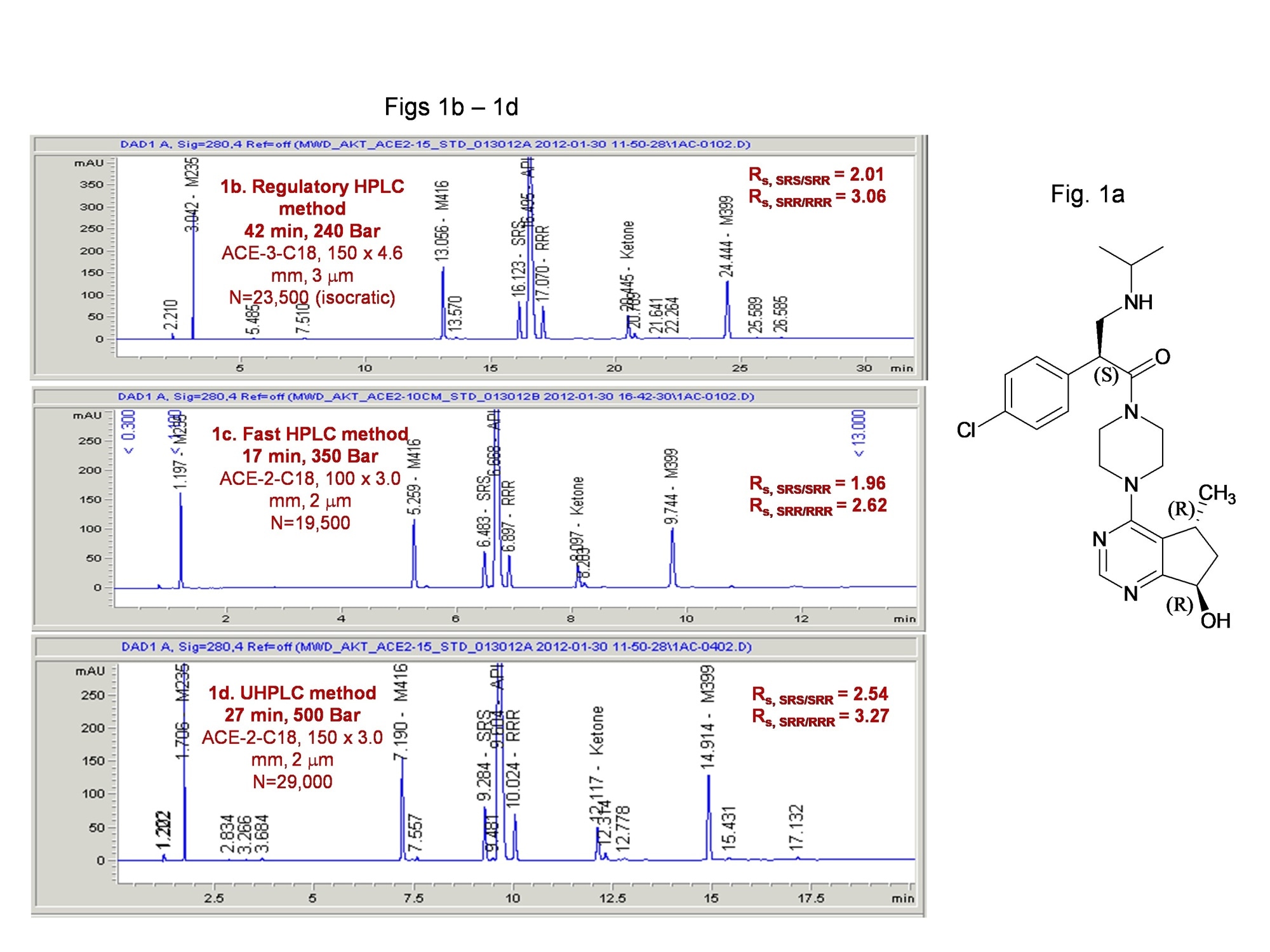
The first one is a HPLC stability-indicating method for an API with 3 chiral centers mentioned before. Figure 1a is the structure of the NCE; 1b is the regulatory HPLC method, while 1c and 1d are UHPLC methods used to support phase 2 drug product development which are faster and has higher resolution.
The second case study illustrates the high-resolution capability of a well-developed UHPLC method for a drug product with 4 APIs using a 5-gradient-segment method capable of separating up to 60+ impurities in a robust manner.

Please give a brief introduction to the talk you will be presenting at Pittcon 2019.
In this upcoming presentation at Pittcon 2019, I will discuss several approaches to expedite HPLC method development using UHPLC and an a 3-pronged method template approach with a focus on stability-indicating analysis of complex pharmaceuticals.
Where can readers find more information?
- M. W. Dong and K. Zhang, UHPLC in method development, Trend in Anal. Chem. 63, 21-30, 2014.
- M.W. Dong, A Three-Pronged Template Approach for Rapid HPLC Method Development, LCGC North Am. 31(8), 612-621, 2013.
- M. W. Dong, A Universal Reversed-Phase HPLC Method for Pharmaceutical Analysis, LCGC North Am. 34(6), 408-419, 2016.
- M. W. Dong, D. Guillarme, D. Prudhomme, et al., High-resolution separations of complex pharmaceuticals by UHPLC: Case studies and quality control implications, LC GC North Am. 32(11), 868-76, 2014.
- D. Guillarme and M. W. Dong (Eds). UHPLC: Where we are ten years after its commercial introduction, Trends in Anal. Chem., 63, 1-188, 2014 (Special issue).
About Dr. Michael Dong

Dr. Michael W. Dong is a principal consultant in MWD Consulting focusing on consulting and training services on HPLC/UHPLC, pharmaceutical analysis, and drug quality. He was formerly Senior Scientist in Analytical Chemistry and Quality Control at Genentech, Research Fellow at Purdue Pharma, and Senior Staff Scientist at Applied Biosystems / Perkin-Elmer.
He holds a Ph.D. in Analytical Chemistry from the City University of New York, and a certificate in Biotechnology from U. C. Santa Cruz. He has 120+ publications including a bestselling book on chromatography (Modern HPLC for Practicing Scientists, Wiley). He is an editorial advisory board member of LCGC magazine, American Pharmaceutical Review, and Chinese American Chromatography Association.
About Pittcon
Pittcon is North America’s largest conference and exposition in laboratory science, attracting thousands of industry, academia, and government attendees and exhibitors from around the world in. Pittcon goes beyond the typical conference; it's a global community dedicated to advancing science through education, collaboration, and philanthropy. By bringing together scientists, researchers, educators, students, and industry leaders, Pittcon creates opportunities to learn, connect, and inspire innovation. Beyond the event itself, more than 90% of Pittcon's net proceeds support science education and outreach initiatives locally and globally, funding scholarships, grants, STEM programs, laboratory improvements, and other efforts that strengthen communities and expand access to science.
Pittcon delivers world-class educational and professional development opportunities through its Technical Program, Short Courses, and Networking Workshops and Roundtables. Attendees can explore the latest scientific research through symposia, oral and poster presentations, enhance their skills with expert-led deep-dive courses, and connect with peers through interactive workshops and networking experiences designed to foster collaboration and career growth.
The Pittcon Expo brings the scientific community together in an immersive environment where attendees can discover the latest laboratory technologies, meet with leading instrument and service providers, and experience science in action. Beyond the exhibit booths, the Expo Floor features engaging attractions, educational experiences, demonstrations, and networking spaces, while receptions, parties, and social events create opportunities to build lasting connections with colleagues from around the world.
 Synthetic Psychedelics: A Growing Public Health Concern
Synthetic Psychedelics: A Growing Public Health Concern