This article and associated images are based on a poster originally authored by Lin Chen, Vincent Blay, Pedro J. Ballester and Douglas R. Houston and presented at ELRIG Drug Discovery 2025 in affiliation with University of Edinburgh, University of California at Santa Cruz and Imperial College London.
This poster is being hosted on this website in its raw form, without modifications. It has not undergone peer review but has been reviewed to meet AZoNetwork's editorial quality standards. The information contained is for informational purposes only and should not be considered validated by independent peer assessment.

Introduction
The discovery of effective therapeutics remains a complex, costly, and time-consuming endeavor. A central bottleneck in early-stage drug discovery is identifying suitable hit compounds with moderate affinity for known biological targets. The research team present SCORCH2, a machine-learning framework to enhance virtual screening performance by using interaction features.
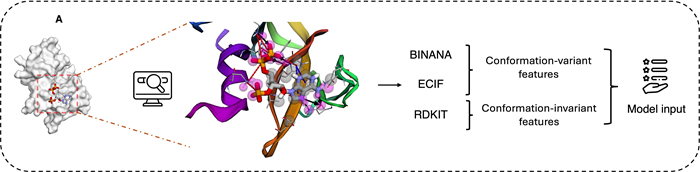
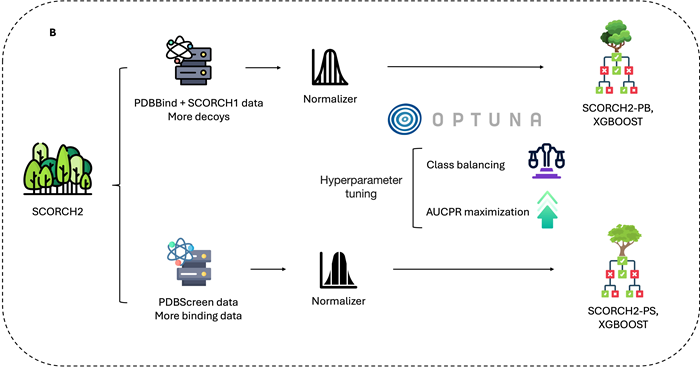
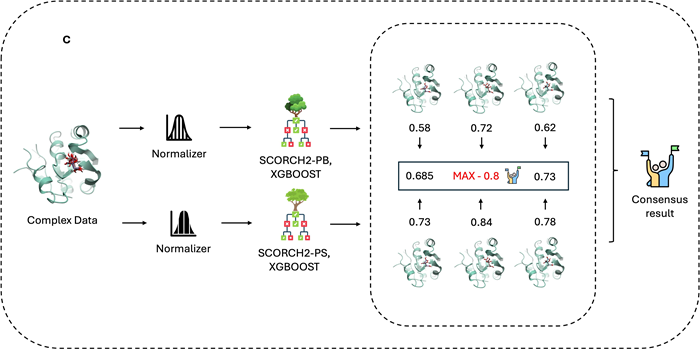
Figure 1. A) Molecular interaction visualization and feature combination. Crystal structure of protein with PDB ID: 1AFK, highlighting interactions with the ligand PAP (3′-Phosphate-Adenosine-5′-Diphosphate). Fuchsia spheres represent atoms in closest contact with the ligand (cutoff within 2.5 Å). Black arrows indicate hydrogen bonds, red dashed lines represent salt bridges, and blue dashed lines denote π–π stacking interactions. SCORCH2 features are primarily generated from three methods: BINANA, ECIF, and RDKit. While BINANA and ECIF extract conformation-sensitive features, RDKit provides features that remain independent of conformational changes. B) SCORCH2 structure and model training. SCORCH2 uses a simplified architecture consisting of two XGBoost models. Each model is trained with different data, and Optuna is used for optimal parameter search with appropriate training strategies. C) SCORCH2 inference workflow, for the complex data, these models are designed to operate independently, and the final result is provided by a weighted consensus. Image Credit: Image courtesy of Lin Chen et al., in partnership with ELRIG (UK) Ltd.
Methods
SCORCH2 uses two distinct XGBoost1 models trained on separate datasets for heterogeneous consensus scoring. Hyperparameter optimization was conducted using Optuna2, targeting AUCPR maximization for imbalanced datasets.
For the inference, the team used a weighted consensus derived from the maximum-scoring pose. They also introduced the concept of Knowledge Pattern (KP) and demonstrated that incorporating diverse KPs can potentially enhance the overall performance.
Results
SCORCH2 outperforms previous docking and rescoring methods on the full DEKOIS 2.0 benchmark. It also exhibits strong robustness on a subset with unseen targets, highlighting its generalization capability.
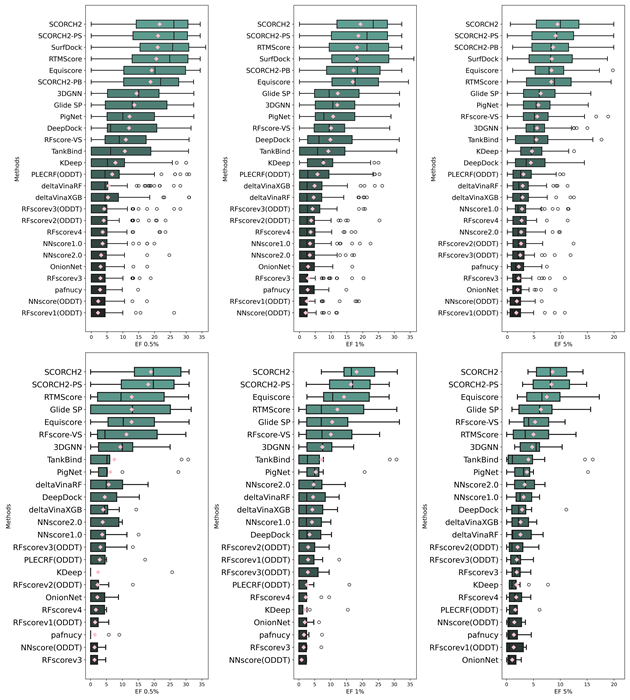
Figure 2. Comparative analysis of virtual screening performance using the complete DEKOIS 2.0 benchmark against other methods (Figure 2, panels A–C), as well as on a subset of unseen targets (Figure 2, panels D–F). The Enrichment Factor (EF) measures the model’s ability to prioritize active compounds over random selection. Image Credit: Image courtesy of Lin Chen et al., in partnership with ELRIG (UK) Ltd.
Model explainability
SHAP-based analysis of two validated Spleen Tyrosine Kinase ligands demonstrates the model’s interpretability and its ability to prioritize critical protein-ligand interactions.
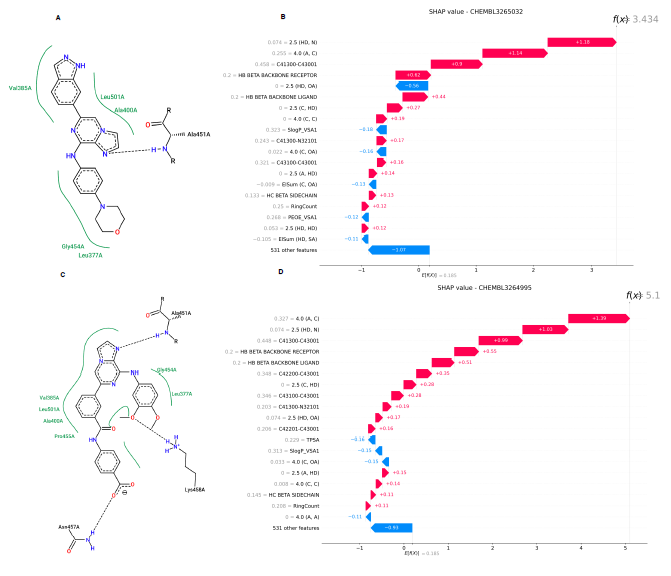
Figure 3. Analysis of molecular interactions and feature importance for two SYK inhibitors using 2D interaction diagrams and SHAP waterfall plots. (A; B) Analysis for CHEMBL3265032: 2D interaction diagram (A) and SHAP waterfall plot (B). (C; D) Analysis for CHEMBL3264995: 2D interaction diagram (C) and SHAP waterfall plot (D). In the SHAP waterfall plots, the color of each bar indicates the value of the corresponding feature (blue for lower values and red for higher values). Features are ranked by their contribution to the model’s prediction, with the most important feature at the top. Image Credit: Image courtesy of Lin Chen et al., in partnership with ELRIG (UK) Ltd.
Conclusion
SCORCH2 underscores the importance of modeling general molecular interactions in virtual screening and provides an effective tool for streamlined prediction. Its strong performance demonstrates that tree-based methods remain competitive in specific scenarios.
Acknowledgement
This research was supported by a Medical Research Council programme grant (MR/Y013131/1) and Rosetrees Trust. L.C. was supported by the Darwin Trust, University of Edinburgh.
References
- Chen, T. and Guestrin, C. (2016). XGBoost: a Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining - KDD ’16, 1(1), pp.785–794. https://doi.org/10.1145/2939672.2939785.
- Akiba, T., et al. (2019). Optuna: A Next-generation Hyperparameter Optimization Framework. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. https://doi.org/10.1145/3292500.3330701.
- Bauer, M.R., et al. (2013). Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0 – A Public Library of Challenging Docking Benchmark Sets. Journal of Chemical Information and Modeling, 53(6), pp.1447–1462. https://doi.org/10.1021/ci400115b.
- Lundberg, S.M., et al. (2020). From local explanations to global understanding with explainable AI for trees. Nature Machine Intelligence, 2(1), pp.56–67. https://doi.org/10.1038/s42256-019-0138-9.
About the University of Edinburgh
Founded in 1583, the University of Edinburgh is one of the oldest universities in the English-speaking world and a major public research university in Scotland.
The university is a member of the Russell Group and other major international research alliances, reflecting its commitment to high-impact research, innovation, and academic excellence.
Among its strengths are the sciences (primarily biological and medical research), humanities, and interdisciplinary fields, with research output that consistently ranks amongst the UK’s top tier.
About ELRIG (UK) Ltd.

The European Laboratory Research & Innovation Group (ELRIG) is a leading European not-for-profit organization that exists to provide outstanding scientific content to the life science community. The foundation of the organization is based on the use and application of automation, robotics and instrumentation in life science laboratories, but over time, we have evolved to respond to the needs of biopharma by developing scientific programmes that focus on cutting-edge research areas that have the potential to revolutionize drug discovery.
Comprised of a global community of over 12,000 life science professionals, participating in our events, whether it be at one of our scientific conferences or one of our networking meetings, will enable any of our community to exchange information, within disciplines and across academic and biopharmaceutical organizations, on an open access basis, as all our events are free-of-charge to attend!
Our values
Our values are to always ensure the highest quality of content and that content will be made readily accessible to all, and that we will always be an inclusive organization, serving a diverse scientific network. In addition, ELRIG will always be a volunteer led organization, run by and for the life sciences community, on a not-for-profit basis.
Our purpose
ELRIG is a company whose purpose is to bring the life science and drug discovery communities together to learn, share, connect, innovate and collaborate, on an open access basis. We achieve this through the provision of world class conferences, networking events, webinars and digital content.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.
Last Updated: Nov 13, 2025