During the process of research and development of antibody drug molecules, an essential step is determining the sequence of the antibody. Mass spectrometry technologies have advanced so far as to make them the first choice in sequence analysis. This article describes three technologies which are frequently chosen for this purpose, namely:
- Intact mass analysis
- Peptide mapping
- Protein de novo sequencing
While intact mass analysis and peptide mapping need the amino acid sequence of the antibody to be known beforehand, the third technique permits sequencing to be done directly using the LC-MS/MS data.
Intact Mass Analysis
Intact mass analysis is a technique which measures the molecular weight of the intact antibody protein, either in native form or as a separate determination of the heavy and light chain once the molecule is reduced. Antibody fragments may also be measured, such as for Fab or VH+CH1.
The determination of the primary sequence of the protein antibody is confirmed by finding the intact mass in this way. However, this does not permit variations in the sequence - such as leucine/isoleucine mutations or amino acid swapping - to be verified. Another use of this technique is to help evaluate the relative ratios of the glycosylated forms which have been expressed, because these modified forms are often bestowed with immunogenicity or biological activity by the sugar residues.
The procedure of an intact mass analysis is typically as below:
- An LC-MS is carried out on the purified antibody, either in intact or fragment form, to obtain a full MS spectral readout
- The high-charged full MS spectrum is deconvoluted
- The data is analyzed and interpreted
The process is depicted in the following image which shows the target protein B with a high-charge envelope and the associated deconvolution spectrum C:
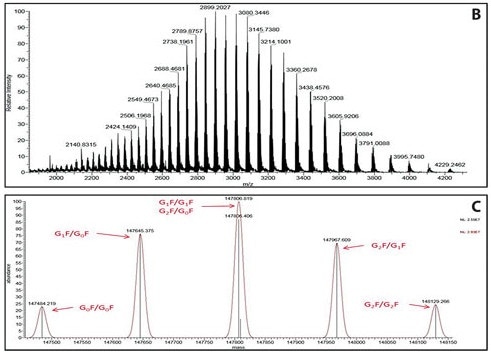
(Image credit: https://www.genengnews.com/insights/analysis-of-ptms-via-capillary-lc-coupled-with-hr-am-mass-spec/)
Peptide Mapping
The technique of peptide mapping uses mass spectrometry to examine peptides obtained by protein digestion. It is based upon comparison and is primarily used to verify the identity of proteins. The requirements are a reference in the form of a reference sequence, a reference standard or a reference material, which can be matched against the peptides of interest to obtain the degree of similarity or identity.
One especially important use is to identify the differences between two or more liquid chromatographic samples or conditions. Another is validating the sequence of a target protein using a known sequence, while a third is to discover point mutations. It is based upon more information than intact mass analysis and yields more reliable results accordingly. It is not suitable to detect hard-to-find mutations such as amino acid swapping.
The general procedure is as follows:
- Monoclonal antibody digestion to generate peptides with the use of one or more enzymes which cleave the protein at different sites
- Peptide separation by chromatographic techniques, such as reverse-phase high performance liquid chromatography (HPLC)
- Using a suitable database search engine (such as Mascot or SEQUEST for MS/MS), the peptides are identified
- The peptides are mapped to the LC chromatogram or to a protein sequence which is already known, and the analysis is thus reported and mapped
A typical tryptic peptide is mapped in the image below using a comparison between three monoclonal antibodies.
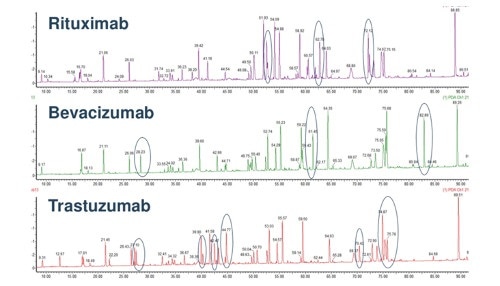
(Image credit: http://www.slideshare.net/pscad123/usp-biotherapeutics-biological-medicines)
The second image below depicts the verification of sequence information by mapping peptides to an already determined protein sequence.
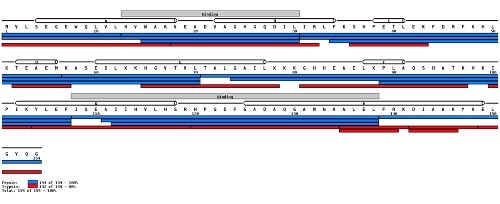
(Image credit: https://peterslab.org/MSTools/)
Protein De Novo Sequencing
The technology of de novo sequencing of proteins is meant to determine the antibody protein sequence directly using mass spectrometry without the need for knowing the sequence of the protein beforehand. A high mass accuracy instrument is used to measure the peptides and to derive the sequence of the protein from the overlapping peptides and fragment ion information.
The basis of protein de novo sequencing is an algorithm for peptide de novo which generates de novo peptide sequences and scores individual amino acids with precision. This is of great value when it comes to determining the CDR (complementarity-determining region) of the antibody, of which there are six on the light and heavy chains.
These are highly variable regions, and are responsible for the diversity upon which the immune system function is based. However, this same variability introduces unreliability into every known database of sequences for these regions.
The procedure is as follows:
- The digestion of monoclonal antibody to peptides using one or more enzymes which act on different cleavage sites
- Peptide separation with reverse-phase HPLC or other chromatographic technologies
- Using a mass spectrometer with high mass accuracy, a tandem mass spectrum is generated
- De novo peptides are generated using peptide de novo sequencing
- The de novo peptides are now reassembled to obtain the full antibody sequence
The image below shows how the antibody sequencing software from Rapid Novor identifies the peptides in a protein sequenced de novo. The bold font captions show those amino acids which are validated by the MS/MS fragment ion peaks. The peptides which overlap are a collective verification that each amino acid in the antibody has been correctly identified.
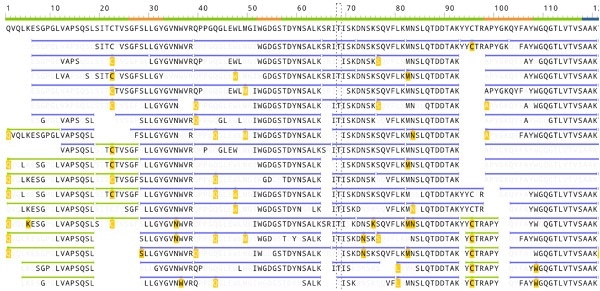
(Image credit: Screenshot of Rapid Novor antibody sequencing software)
Conclusion
While each of the three technologies are of use in different ways, it becomes more complex to carry out experiments and analyze the data from the first to the third. This of course means that the level of expertise and the costs also go up.
However, if the primary sequence is well-known, and the main objective is to prevent the occurrence of variations in the sequence that could lead to significant changes in the protein intact mass, or changes in the mass or retention time of certain peptides, the intact mass analysis and peptide mapping are probably the preferred choices, as they are not only less expensive but produce faster results. However, if each amino acid is to be determined with absolute confidence, protein de novo sequencing becomes the tool of choice.
 Rapid Novor Inc
Rapid Novor Inc
Rapid Novor Inc. is the world's leader in antibody protein sequencing technology. Specializing in the field of mass spectrometry-based proteomics, the team has developed the technology to directly sequence antibody proteins without needing to access the producing cell line.
As a University of Waterloo spin-off, the company is building its technology portfolios based on the twenty years of scientific research and inventions from Dr. Bin Ma, co-founder and chief scientist. The company's REmAb™ antibody protein sequencing service has allowed for the accurate sequencing of any given antibody proteins on a routine basis. The company's WILD™ service is the first commercially available service that can accurately distinguish the isomeric Isoleucine and Leucine using mass spectrometry.
Rapid Novor Inc's mission is to advance life science for better human health with next generation protein sequencing.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.