In this interview, Ghaith Hamza from AstraZeneca talks to News-Medical Life Sciences about the basic principles of Affinity-Bead Assisted Mass Spectrometry (Affi-BAMS) and how Affi-BAMS can help target specific proteins for proteomic applications.
Please give an overview of AstraZeneca’s work and the main areas you are involved in?
AstraZeneca’s main therapeutic areas revolve around oncology, cardiovascular, renal, metabolism, and respiratory therapies. However, we are also working on opportunity-led projects focused on autoimmunity, infectious diseases, and neuroscience.
AstraZeneca’s R&D department focuses on 5 ‘R frameworks’. These 5 Rs are: identifying the right target, making sure our molecules get to the right tissue, ensuring the right safety approach with minimal side effects, and selecting the right patients - ones that will benefit the most from our drugs - and, of course, defining the right commercial value and ensuring future viability. These principles are underpinned by the ‘right culture’ - truth-seeking behaviors and scientific rigor.
Can you give our readers some examples of the different groups and projects within AstraZeneca?
The Discovery Science Department operates within the R&D department, and within that, we have the Chemical Biology and Proteomics group - this is the group that I currently work in. Within this group, we focus on three main pillars of work: target discovery, multiOMICs, and protein degradation.
Our target discovery work focuses on target deconvolution, target engagement, selectivity, and off-target profiling. To facilitate this, we utilize a suite of assays to characterize our targets, ranging from chemical probes to family affinity matrices, cellular thermal shift assays, and global proteomics.
Our multiOMICs work focuses on clinical translation, harnessing the power of large scale quantitative clinical proteomics. Here, we integrate data sets with multiple OMICs technologies to better understand disease mechanisms; characterizing preclinical models and clinical patients’ samples in order to better stratify the patients, as well as our preclinical models.
Lastly, the protein degradation group focuses on target validation and therapeutic applications. Here, we utilize a suite of mass spec binding and degradation assays to focus on protein turnover rate and protein expression while utilizing PROTAC systems.
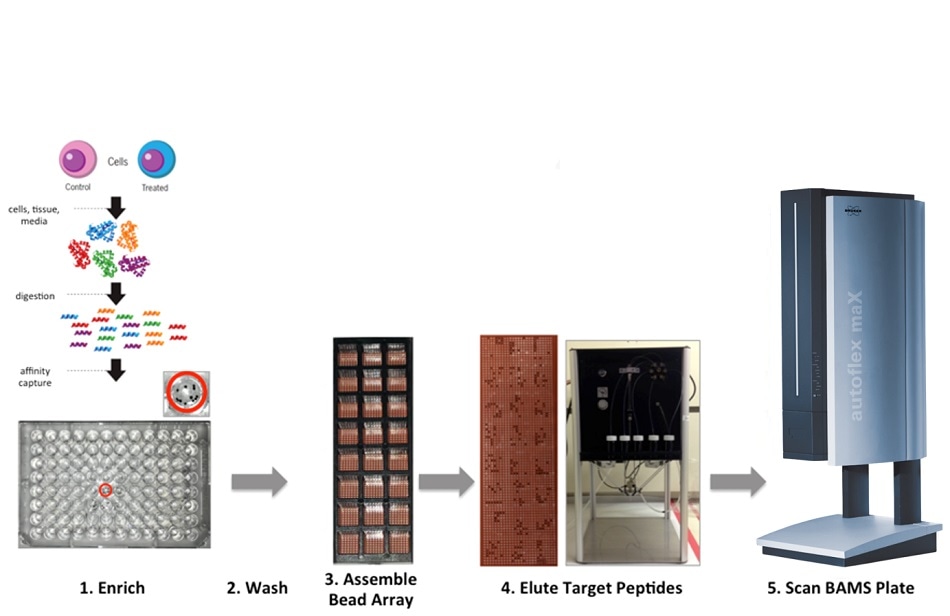
Affinity-Bead Assisted Mass Spectrometry Workflow
What role does Affinity-Bead Assisted Mass Spectrometry play in your work?
Affinity-Bead Assisted Mass Spectrometry (Affi-BAMS) harnesses the power of three main analytical techniques. We use magnetic beads that can simultaneously enrich protein targets within hundreds of samples, and we utilize microarrays to transfer or enrich targets, and this microarray is eluted with a MALDI matrix solution. This then ends up forming a very uniform microarray that can be fed into any mass spectrometer with a MALDI source.
Using this approach, we can generate a single bead on a microarray that captures a single target, and we can transfer this onto a single well. This is effectively a high throughput magnetic bead array platform designed for targeted protein analysis, and we are applying this technique across all three pillars of work within the Chemical Biology and Proteomics group to help us in our R&D efforts.
The Affi-BAMS workflow is very straightforward. We can extract the proteins from any biological sample, whether it be tissue specimens, biofluids, or cells - as long as we can extract their proteins, we can expose them to proteolysis from a variety of different proteases, ultimately generating peptides that contain specific epitopes.
These epitopes can then be targeted through the use of antibodies that are covalently bound to magnetic beads before being arranged on this microarray. Using this approach, we can form a very uniform microarray that can be fed into a MALDI-TOF instrument for clean retrieval and quantitation.
Affinity-Bead Assisted Mass Spectrometry - Ghaith Hamza
What is the specific elution process used here?
There are two key elements of this process. An elastomer is used to confine each single bead to each well. These confined beads are then exposed to a highly organic and highly acidic solution containing a MALDI matrix. It is then possible to elute our targets and confine these eluted target peptides within the well. The matrix crystallizes, forming very uniform, tiny spots.
Essentially, we can eliminate the irregular crystal formations that naturally occur in MALDI matrices. Each probe is validated upfront, once. As the antibody and the enrichment are validated, it is possible to subsequently acquire the MALDI-TOF spectra.
What are some of the most typical applications and advantages of Affi-BAMS?
Affi-BAMS is a targeted platform that can be utilized in many different assays or applications. One such example is SILAC quantitation, and one example study utilized two kinase inhibitors: Staurosporine and SU11274. The target of interest in this example was C-MET, and investigations discovered that there was a significant decrease in phosphorylation states under the SU treatment while a modest decrease was observed under the ST treatment – essentially, the Staurosporine compared to the control. This was found to be in line with a Western blot of the same experiment.
A key advantage of Affi-BAMS is that it utilizes mass as the second layer of quantitation. It is ideal for applications that have multiple modifications (such as the case in RPS6 on its c-terminal tail, where it is heavily phosphorylated), as these conditions would not be visible in a regular Western blot. However, because the quantitation is resolved based on mass, it is possible to detect and quantitate each phosphorylation species on a sample including an RPS6 peptide.
It is also possible to undertake a very targeted pathway analysis. Another example – a SILAC experiment on MKN45 cells treated with rapamycin – involved an mTOR target. This example saw the use of a multiplexed assay in which each bead enriches a single target, but we were able to quantitate each single target within that assay.
Here, mTOR phosphorylation was found to decrease, and as well as the downstream targets of mTOR 4EBP and RPS6, the phosphorylation status also decreased dramatically. The AKT levels were especially interesting, and as the total expression increased, while 4EBP increased slightly. When examining a T37 & T46 on 4EBP, we saw a dramatic decrease in the doubly phosphorylated form, while in the singly phosphorylated form, we saw a slight increase. This is a good example of the way in which we can set up multiplexed assays for your pathway of choice while being able to intricately dissect the biology.

Prepared BAMS™ Microarray Slide
How useful is Affi-BAMS when working with absolute concentrations?
Very useful. We can work with absolute concentrations via the use of SISCAPA Quantitation. We recently completed a study of biofluid plasma, and we designed this study by generating two curves - the forward and reverse curve. In the forward curve, we varied the light concentration, and we fixed the heavy peptide concentration. Meanwhile, in the reverse curve, we varied the heavy peptide concentration and fixed the light concentration.
When we analyzed the results, the reverse curve showed us the dynamic range of the assay for this specific peptide spanning many orders of magnitude, as well as the limit of detection and limit of quantitation. The forward curve demonstrated the analyte concentration.
What are the benefits of specifically using targeted assays?
Using a targeted assay, we can target proteins of interest and look for the sort of point mutations that frequently arise in drug resistance, and we can target these specific proteins with point mutations, monitoring how this translates to drug resistance.
Again, because this is a targeted platform, we can look for peptides generated within the biological system by different enzymes or sheddases, effectively monitoring different sheddase activity. For example, when working with a beta-amyloid from undigested cerebral spinal fluid, we can effectively utilize bead multiplexing because the epitope is retained within the range of different peptide fragments generated. We can cleanly retrieve many of the fragments, segregating between the disease and healthy samples. We can use this technique to understand how this process translates to relevant biology.
BAMS™ Assay Kits
Can this approach also be used within epigenetics?
We can use this approach when working with epigenetics for specific histones. For example, histone 3 is very heavily modified using a variety of post-translational modifications. In a recent study, we looked at a U2OS cell line that had been treated with an HDAC inhibitor. We increased acetylation events on histones, digested the control samples and treated samples with chymotrypsin, ultimately generating an interesting mass spectrum.
Using this spectrum, we could focus on one peak and use this as a reference peak to normalize between the two samples. From there, we examined several peaks, sequencing two of them. Finally, we looked at the post-translational modification crosstalk occurring on histones and translated that into a phenotype (a biological readout) to see how epigenetic modifications on histones actually translate into biology.
In another epigenetics application, we examined brain tissue that had been digested with LysC. A range of different peptide fragments were retrieved including, interestingly, peptide fragments that correspond to tail clipping events. We were able to detect two acetylation events, as well as mono-, di-, and trimethyl in this peptide fragment.
Being able to detect both the peptide generated by the LysC condition and the peptide generated by tail clipping events, happening within the tissue itself, is an important step in better understanding biology, and translating this information into a phenotype to effectively monitor the tail clipping and biological events.
About Ghaith Hamza
.jpg)
Ghaith Hamza is a Senior Proteomics Scientist at AstraZeneca studying Mass Spectrometry, Proteomics, Genomics & Chemical Biology to investigate human diseases and understand the mechanisms of cellular signaling pathways.
 Single-cell proteomics – when mass spectrometry revolutionizes biomedical research
Single-cell proteomics – when mass spectrometry revolutionizes biomedical research