Multi-dimensional separations are essential to obtain a precise and detailed view of the content of complex proteomics samples in both targeted (quantitative) and discovery workflows, and since the 1960s, ion mobility has been used as an extra separation device in commercial mass spectrometers.
In recent times, trapped ion mobility spectrometry (TIMS) has been launched and combined with QTOF mass spectrometers. In TIMS, ions are driven via a TIMS tunnel by a gas flow. An electric field catches each ion at the position where the push that it experiences from the gas flow corresponds with the force of the electric field, leading to separation by mobility, charge in the gas phase, and a function of their three-dimensional size (Figure 1).
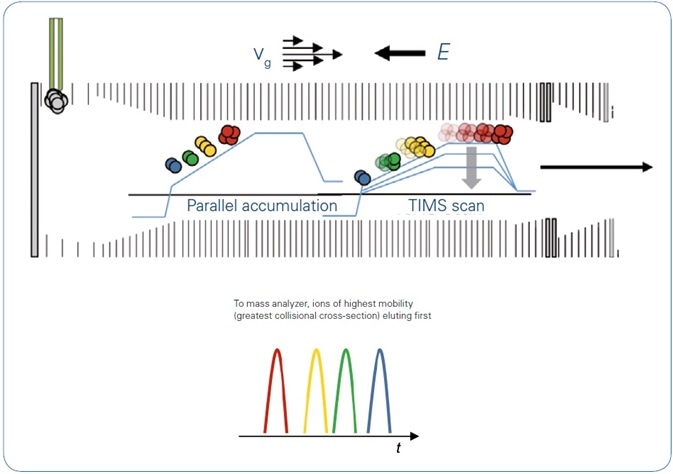
Figure 1. TIMS operation mode: With the directed forces of gas flow, vg, and electrical field, E, ions are accumulated and trapped according to mobility. Step-wise decrease of the electrical field permits serial elution of separated ions, which are then transferred to the QTOF. Using the parallel accumulation operation mode ions are eluted from the second part of the TIMS device while the next series of ions is introduced into the first part of the TIMS device.
When the electrical field is ramped down, selective ions are released from the TIMS tunnel based on their ion mobility (Ω/z), where Ω is the collisional cross-section (CCS) of the ion. Contrary to conventional drift tube separation by size-to-charge, the larger ions elute initially, followed by ions of decreasing cross-section. In this research, a timsTOF Pro instrument (Bruker Daltonics) was employed, with the eluted ions further analyzed through quadrupole time-of-flight mass spectrometry (Figure 2).
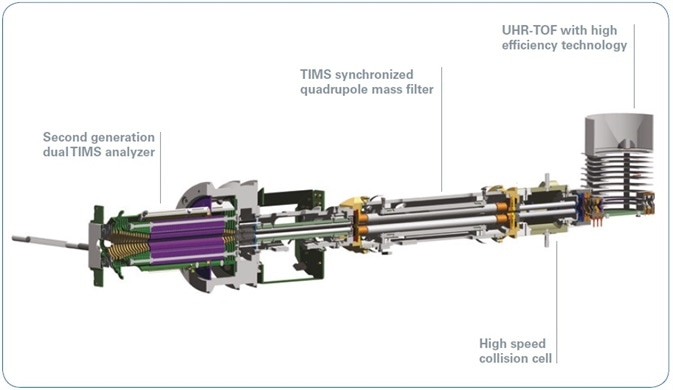
Figure 2. Ion optics of the timsTOF Pro instrument including a dual TIMS analyzer and QTOF mass spectrometer.
The spectra produced at the time of the TIMS elution cycle are called TIMS scan. The two-dimensional separation of ions (m/z and 1/K0) - summed across a TIMS scan - can be visualized as a TIMS MS heat map, with m/z and mobility as the x and y axes, respectively (see Figure 3).
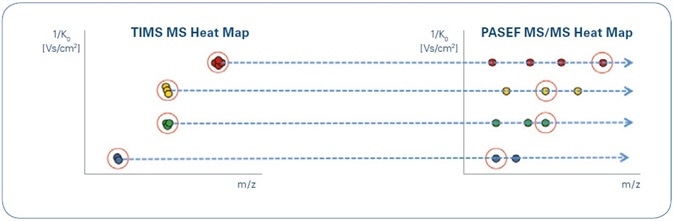
Figure 3. Illustration of TIMS MS (left) and PASEF MS/MS (right) heat maps resulting from TIMS separations. Although the data generated may be very complex, the characteristic ion mobility of targeted precursors and their fragments facilitates easy data alignment. Note that precursors of the same m/ z (yellow and green) may be clearly separated by their mobility.
Similarly, specific ions may be isolated in three dimensions based on their TIMS elution time, the retention time of liquid chromatography (LC), and m/z (in this research, by a quadrupole filter) and subjected to collision-induced dissociation. In the generated PASEF MS/MS heat map, it is possible to align the fragments with their precursors in the TIMS MS heat map since they occur at the same ion mobility (Figure 3).
Although this orthogonal separation can offer a more in-depth view of complex proteomics samples, its utility for peptide identification may be challenged in various instruments because of poor sensitivity caused by low ion abundance (resulting in weak spectra from successive collision-induced fragmentation), and also high scanning speeds needed to be compatible with the usual 100 ms TIMS scan times. By implementing the “Parallel Accumulation Serial Fragmentation” (PASEF) technique on a rapid scanning QTOF instrument, fragment ion spectra from isolated precursor ions may be obtained more quickly while maintaining spectral quality.
By employing a dual TIMS analyzer, ions may be almost continuously arranged for accumulation, sorting, and elution by mobility, enabling a duty cycle close to 100% (Figure 1). The quadrupole changes its (mass) isolation position a number of times during the period of each IMS scan, allowing the collection of MS/MS spectra from several precursors (see Figure 4).
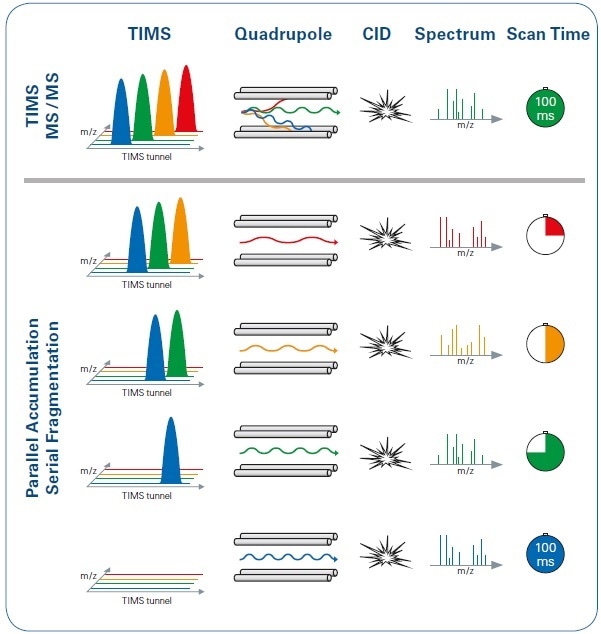
Figure 4. Illustration of the PASEF method in comparison with the standard TIMS MS/MS operation mode, with the same 100 ms timescale. Using PASEF (lower panel), the quadrupole switches its isolation position several times during each ion mobility scan, with multiple precursors isolated and subsequently transferred to the collision cell. In contrast, with the standard TIMS MS/MS approach (upper panel), only one precursor from each TIMS scan is selected.
In addition, spectra for low abundant precursors can be summed to enhance signal intensities to boost the number as well as the confidence of peptide identifications. The application of ion mobility separation before MS/MS results in enhanced signal-to-noise ratios, since the targeted ion species is time focused and later transferred to the collision cell only during the duration of time they elute from the TIMS device. In addition, because of this further dimension of separation, precursors of the same m/z co-eluting from the LC could be distinguished which is impossible with mass-based selection alone.
Two Dimensional (m/z-Mobility) Data Dependent Precursor Acquisition
The need for computational time for determining the precursors that will be chosen for MS/MS is another challenge in the rapid acquisition of data. Maximizing the number of quality MS/MS spectra, a necessary step in successful shotgun proteomics experiments, requires consideration of relative ion intensities and resolutions, the latter of which is increased (thus increasing the number of viable precursors) with the second dimension of separation. Lower intensity targets may be fragmented several times with higher speed data acquisition capabilities. A relatively fast and simple algorithmic method[3] was developed to effectively plan the successive fragmentation scans on the basis of the initial detection of precursor ions at the time of the MS scan in both mobility dimensions and m/z. Computational time does not surpass 1 millisecond even in extremely complex proteomics samples, thus supporting high-throughput data collection quite easily.
Experiment
A complex mixture of tryptic peptide derived from HeLa cells was diluted with 0.1% formic acid (FA) in water to 200 ng/µL concentration. The timsTOF Pro mass spectrometer was connected to a nanoElute UHPLC (Bruker Daltonics). The peptide mixture (200 ng) was transferred to a C18 column (25 cm X 75 µm X 1.6 µm, IonOpticks, Australia). Chromatographic separation was performed using a linear gradient of 2% to 37% of buffer B (0.1% FA in ACN) at a flow rate of 400 nL/minute over 90 minutes. Next, MS data were collected over an MS/MS range of 100 to 1700, and m/z range of 100 to 1700. During the collection of MS/MS data, each TIMS cycle was 1.1 seconds and contained 1 MS + an average of 10 PASEF MS/MS scans. The data, thus obtained, were submitted to the MASCOT search engine for the identification of peptides.
Rapid Data Collection….
As may be anticipated for this kind of sample, the base peak chromatogram (BPC) (see Figure 5, top) signifies a highly complex collection of peptide species. The power of the PASEF technique is easily observed, considering one narrow time slice of the LC gradient (61.7 minutes).
At the time of this example scan, 41 precursors were fed into the scheduling algorithm and addressed by 10 PASEF MS/MS scans, in total. By evaluating one TIMS elution (time) heat map (see Figure 5, bottom left), quick quadrupole isolation window switching for the first 12 precursors becomes obvious. Extra precursors are targeted in successive segments of the elution gradient. Figure 5 (bottom right) signifies all the precursors assessed within this example PASEF cycle, including the re-sequencing of lower abundance targets. The equivalent of 119 separate MS/MS events (if collected without PASEF on a timsTOF mass spectrometer) within this single PASEF cycle (1.1 seconds) were performed.
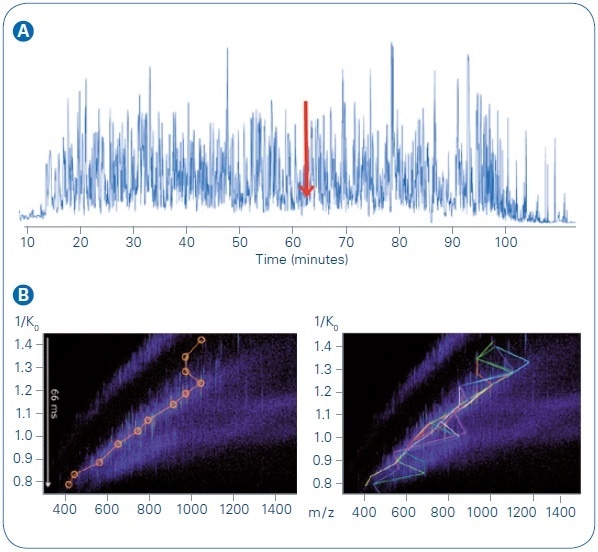
Figure 5. LC MS/MS run, 200 ng HeLa digest, 90 min gradient. A: Base peak chromatogram. B: TIMS MS heat map at 61.7 minute, with targeted precursors indicated. Bottom left, first PASEF MS/MS analysis event, precursors circled in orange. Bottom right, full PASEF MS/MS cycle, with each of the ten colored lines indicating targets from one TIMS elution segment. The most abundant precursors were sequenced in previous scan cycles and were dynamically excluded from re-sequencing.
…While Maintaining Necessary Sensitivity
Significant proteomic features are usually hidden within low-intensity peaks of complex samples, peaks which may not be targeted by instruments that have slower MS/MS acquisition rates. Figure 6 shows the separation power (through differences in ion mobility) and the sensitivity (through the TIMS time focusing and the potential to sum spectra of low abundance targets) of the PASEF method.
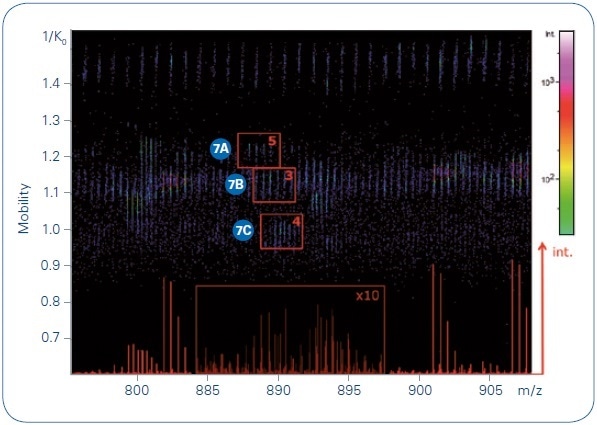
Figure 6. Collection of precursors at m/ z 887.96, 888.94, and 889.78 indicated on a TIMS MS heat map, with the number of summed MS/MS spectra for each indicated. The base peak chromatogram for this narrow m/ z range is shown in the bottom of the heat map.
Summed MS/MS spectra from three low-intensity precursors of analogous m/ z in the spectrum’s dense region were easily detected with low expectation values (probabilities to be a random match), excellent sequence coverage, and high MASCOT scores. Evidently, high performance of the (subsequent) QTOF is equally crucial, and the timsTOF Pro mass spectrometer offers high isotopic fidelity (TIP™ or True Isotopic Pattern), ppm accurate mass, and high resolution. The advantage of this integrated method for shotgun proteomics applications is distinctly shown by the increased confidence of peptide identifications (see Figure 8). In the same sample (200 ng HeLa digest, eluted from the LC with a 90 minute gradient), almost twice as many peptides were found using the PASEF method as compared to analysis with no trapped ion mobility spectrometry separation (using the same instrumentation, yet, with the TIMS device switched off).
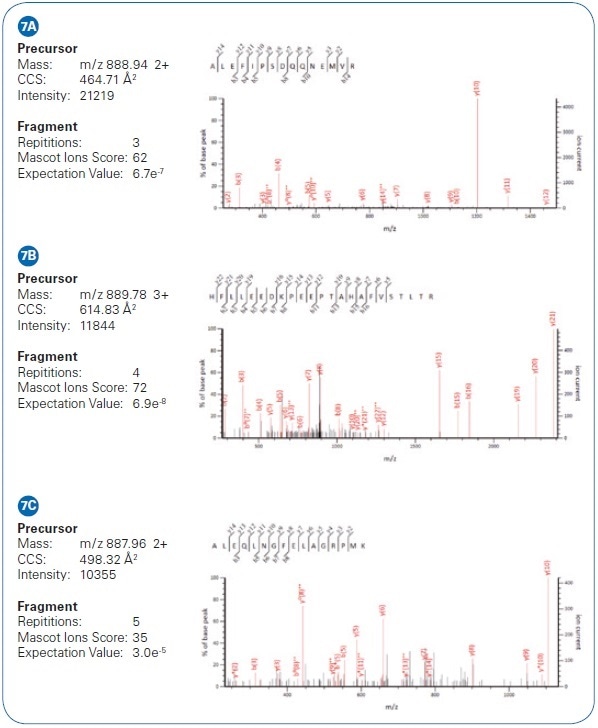
Figure 7. Example of low-intensity peptide matches from HeLa cell analysis via PASEF differ by only 2 Da. While their isotopic patterns overlap in m/ z, they are separated in the mobility dimension and the PASEF MS/MS results yield confident identifications.
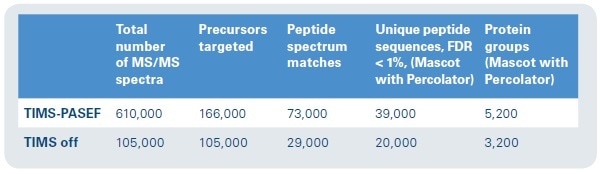
Figure 8. Protein ID results. Approximately six times as many MS/MS spectra can be collected in the same time frame, with nearly double the number of unique peptide sequences and 1.5 times as many protein groups represented.
Discussion
The newly introduced trapped ion mobility spectrometry (TIMS) has added an innovative dimension to quadrupole time-of-flight (QTOF). The integration of the formerly explained “Parallel Accumulation Serial Fragmentation” (PASEF)[1] approach within Bruker’s proprietary timsTOF Pro instrument considerably enhances these advantages. In this experiment, over 160,000 independent precursors from a HeLa digest, isolated using a 90 minute LC gradient, were addressed, with extremely fast MS/MS data acquisition rates.
The timsTOF Pro used for this study offers crucial advances in terms of sensitivity and separation. Trapped ion mobility spectrometry separation alone results in enhanced spectral quality due to the reduction of background noise. Within the TIMS device, the use of two regions of separation allows 100% duty cycle so that no ions are lost, allowing them to accumulate in the first region of the TIMS device while being scanned out into the QTOF from the second region. The timsTOF Pro’s quadrupole can change its isolation window on the millisecond time scale, allowing the QTOF to address more than 10 precursors in a typical 100 msec TIMS scan. The sensitivity is enhanced through the additional time focusing attained by the TIMS, and extra sensitivity for low abundance precursors is realized by addressing the same precursor in multiple PASEF scans.
Table 1 and 2. Instrumental Details
| LC-Settings |
|
| LC-System |
nanoElute UHPLC system (Bruker Daltonics) |
| Column |
25 cm X 75 μm 1.6 μm C18 column (IonOpticks, Australia) |
| LC flow rate |
400 nL/min |
| LC elution conditions |
2% to 37% (100% ACN, 0.1% FA) |
| Column oven (Sonation) temperature |
50°C |
| Source |
CaptiveSpray Ion Source (Bruker Daltonics) |
| Ionization |
ESI (+) |
| MS-Settings |
|
| MS-System |
timsTOF Pro mass spectrometer (Bruker Daltonics) |
| TIMS elution |
100 ms accumulation and ramp time (100% duty cycle) |
| Cycle time |
1.1 s cycle with 1 MS + 10 PASEF MS / MS |
| Scan range |
100 – 1700 m / z |
Conclusions
“Parallel Accumulation Serial Fragmentation” (PASEF) on a timsTOF Pro instrument (Bruker Daltonics) has been demonstrated to effectively deliver considerably higher sensitivity and speed in a data-dependent shotgun proteomics workflow, as shown by the analysis of 166,000 independent precursors from a HeLa digest using a 90 minute gradient. By using PASEF, the number of identifications can possibly be increased to over 40,000 exclusive peptide identifications as well as 5,500 protein identifications from just 200 ng of HeLa digest injected on column.
The number of precursors that can be targeted with this method is radically increased due to the rapid separation of samples by both trapped ion mobility spectrometry and chromatography. These unique acquisition rates also support enhanced sensitivity, since low abundant precursors may be targeted a number of times. This exceptional combination of features offers a new standard for shotgun proteomics performance, allowing researchers to find and detect more biologically pertinent proteins.
References
[1] Meier, et al. (2015) Parallel Accumulation - Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J Proteome Res. 14(12):5378-87
[2] Beck, et al (2015) The Impact II, a Very High-Resolution Quadrupole Time-of-Flight Instrument (QTOF) for Deep Shotgun Proteomics. Mol Cell Proteomics. 14(7):2014-29.
[3] Lubeck et al. Evaluation of a peptide selection algorithm for trapped ion mobility with parallel accumulation serial fragmentation (tims PASEF) on a QTOF instrument. Poster presented at: 65th ASMS Conference on Mass Spectrometry and Allied Topics, June 6, 2017; Indianapolis, IN.
About Bruker Daltonics
Empowering Science – Improving Life
Bruker Daltonics delivers cutting-edge mass spectrometry solutions and workflows that help scientists and industry leaders tackle real-world challenges and make new discoveries. From life sciences and pharmaceutical research to food and contaminant analysis, environmental monitoring, forensics, and industrial quality control, our technologies and instruments provide the precision and reliability you need to make confident decisions.
Our innovative platforms - such as timsTOF, scimaX, neofleX, and DART-TQ - combined with advanced software like SCiLS™ Lab, MetaboScape®, and Biopharma Compass®, transform complex data into actionable insights. Breakthrough innovations like Trapped Ion Mobility (TIMS), Omnitrap®, and dual ionization GC-HRMS are redefining what’s possible in mass spectrometry.
Trusted by leading research institutes, universities, government agencies, and industrial partners worldwide, Bruker Daltonics is committed to driving scientific progress and delivering solutions that matter.
(For Research Use Only. Not for use in clinical diagnostic procedures).
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.