The structure of biopharmaceuticals can be altered by chemical artifacts like disulfide structure modification or deamidation events. In this context, the formation of trisulfides is a particularly challenging artifact.
During the development of antibody drug conjugates, high trisulfide levels tend to interfere with specific conjugation chemistries. Such potential problems should be addressed early in the development pipeline in order to prevent costly remediation downstream.
| Keywords |
Instrumentation and Software |
| Biopharmaceuticals |
rapifleX MALDI-TOF/TOF |
| PTMs |
ultrafleXtreme MALDI-TOF/TOF |
| Artifactual modifications |
DisulfideDetect software |
| Disulfide Crosslinks |
LC-MALDI Fraction Spotter FCII |
| Scrambling |
|
| Trisulfides |
|
Therefore, a fully automated technique has been already developed for identifying disulfide structures in biopharmaceuticals predicated on an LC-MALDI-TOF/TOF workflow. In this analysis, the extension of this workflow was assessed in relation to the extra detection of trisulfides.
Method
In the experimental method, a monoclonal IgG1 antibody (mAb) sample with increased levels of trisulfide formation (Pfizer) and a range of mAbs - IgG1, IgG2, and IgG4: adalimumab, panitumumab, and natalizumab - available on the market were digested using Lys-C/trypsin under non-reducing conditions. nanoLC was used to separate peptides, which were detected on a MALDI target at an interval of 10 seconds. MS/MS spectra were subsequently acquired using a rapifleX MALDI-TOF/TOF at high speed - utilizing Bruker’s 10 kHz smartbeam 3D laser.
The Bruker DisulfideDetect software - provided for detecting and identifying the anticipated disulfide bonds inclusive of scrambled disulfides - enabled fully automated data analysis. The following manual analysis of the same LC-MALDI dataset produced species 32 Da higher in comparison with the corresponding disulfide - comprising of peptides (DSB-peptides). It was inferred that such species from the detected peptides contained trisulfides (TSB-peptides). For a relatively simpler and quicker analysis, a new technique was developed using stepwise elution from ZipTips (Millipore) to establish the trisulfide content in one MALDI mass spectrum.
Results
Automated disulfide bond analysis using DisulfideDetect software and rapifleX instrumentation established the mAbs’ disulfide bond state, as indicated in Figure 1 for adalimumab.
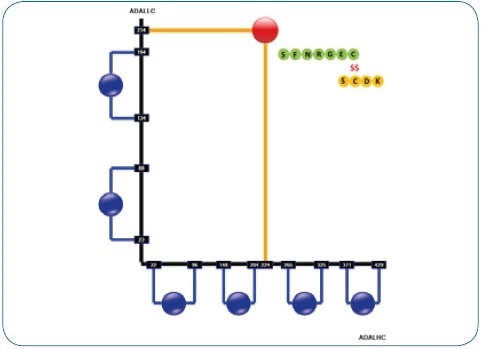
Figure 1. Resulting disulfide structure printout of a human IgG1 antibody (adalimumab) in the DisulfideDetect software. The disulfide bond is highlighted that links LC and HC. This particular site - together with the hinge di-peptide - is susceptible to trisulfide formation.
This method was used to analyze all asymmetric dipeptides with a single disulfide bond. It is known that the inter-chain disulfides between the HC and the LC are particularly sensitive to the formation of trisulfide and were further assessed manually. This peptide present in human IgG1 is a short, hydrophilic one (1261.5 Da), eluting at low acetonitrile concentration in an RP-LC gradient. The LC–HC dipeptide in human IgG2 and IgG4 is larger (2039.94 Da) and elutes much later in the gradient. In all the four antibodies, MS/MS was used to detect and identify trisulfide dipeptides. Table 1 lists the quantitative results.
Table 1. Quantitation results on the LC-HC dipeptides of a Pfizer mAb and other IgG1, IgG2 and IgG4 antibodies.
| mAb |
Method |
Trisulfide content |
| Pfizer IgG1 mAb |
LC-MALDI |
13.5 ± 2.6% |
| Pfizer IgG1 mAb |
ZipTip |
9.5 ± 0.75% |
| Pfizer IgG1 mAb |
ESI-qTOF |
11.3% |
| Adalimumab |
LC-MALDI |
0.6% |
| Adalimumab |
ZipTip |
0.65% |
| Panitumumab |
LC-MALDI |
1.9% |
| Natalizumab |
LC-MALDI |
0.4% |
It was observed that the IgG1 mAb (Pfizer) exhibited an increased formation of trisulfides in the LC-HC dipeptide (see Figure 2) - 11.3% correlates with earlier LC-ESI-qTOF experiments (see Table 1). Three LC-MALDI simulates with three MS measurements each showed an average trisulfide amount of 13.5% ± 2.6% (CV=19%). For the three therapeutic mAbs, trisulfide rates were determined between 0.4% and 1.9% from just one LC-MALDI experiment.
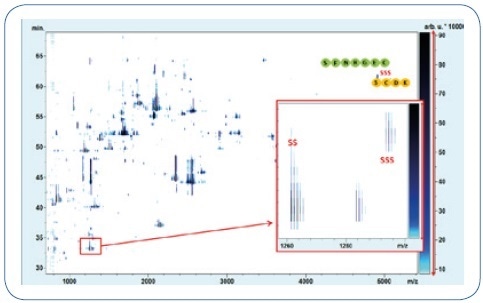
Figure 2. LC-MALDI-MS analysis of the IgG1 mAb (Pfizer) reveals increased disulfide formation. The regular LC-HC linkage DSB-peptide (SS) and the corresponding TSB-peptide (SSS) are marked accordingly. The disulfide and disulfide dipeptides elute with a difference of 2 minutes and were identified by MALDI-TOF-MS/MS.
Apart from the LC-MALDI analysis, a simple technique was also developed to rapidly check, for example, in screening situations - the trisulfide formation of LC-HC dipeptide in human IgG1 antibodies. Furthermore, the Lys-C digest was fixed to a C18 ZipTip and elution of the LC-HC dipeptides was done using 10% ACN, 0.1% TFA, and additionally examined through MALDI-TOF-MS (see Figure 3). The result obtained from 21 replicates: average trisulfide content is 9.53% ± 0.75% (CV = 8%).
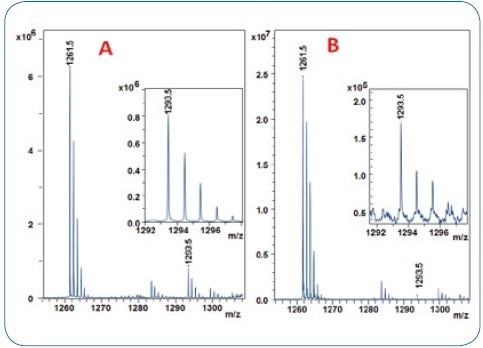
Figure 3. MALDI-TOF analysis of 2 human IgG1 antibody digests using the 10% acetonitrile ZipTip fraction for trisulfide quantitation. A: IgG1 (Pfizer) with 9.5% TSB-peptide content (from Figure2) B: adalimumab with only 0.65 % of TSB-peptide.
MALDI-TOF-MS/MS identified the dipeptides in both - LC-MALDI and ZipTip analyses (see Figure 4).
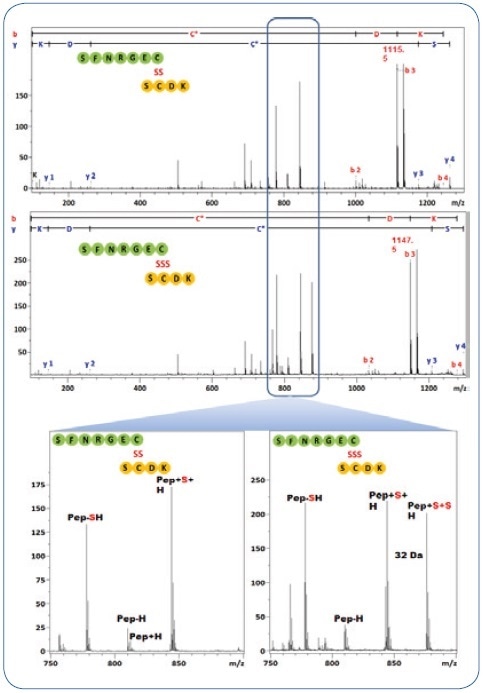
Figure 4. MS/MS spectra from the DSB and TSB peptides in Figure 3. The characteristic fragment pattern for both types of peptides are highlighted, they are used to assign the DSB/TSB peptide-type.
Summary
MALDI-MS was studied as a novel method for identifying and quantifying trisulfides in biopharmaceuticals through:
- An LC-free screening-compatible high-speed approach, which enabled quantification with CV < 10%
- An LC-MALDI-MS/MS technique that produced results similar to LC-QTOF data with CVs < 20%
- Within the rapifleX TOF/TOF, the 10-bit digitizer allowed the quantitation up to three orders dynamic range
The MS/MS data’s quality was extremely high and compatible, enabling an easy detection and identification of trisulfides even without specialized software support.
Conclusions
- Automatic identification of disulfide-bonded peptide
- Trisulfide peptides display a certain fragment ion pattern, which makes it useful to safely detecting them through MALDI-MS/MS
- Trisulfides in mAbs were also manually identified at 32 Da/ +1–3 minutes later in the LC-MS datasets
- Quantification in the 0.1%–1% range was shown (dynamic range ~ 10³)
Acknowledgements
Produced from materials originally authored by Anja Resemann, Rainer Paape, Christoph Nordmann, Waltraud Evers, Lars Vorwerg, Eckhard Belau, Volker Sauerland, Detlev Suckau from Bruker Daltonics Bremen, Germany; Lisa A. Marzilli, Jason Rouse from Pfizer, Andover, MA, USA and Jason S. Wood from Bruker Daltonics, Billerica, MA, USA
About Bruker Daltonics
Empowering Science – Improving Life
Bruker Daltonics delivers cutting-edge mass spectrometry solutions and workflows that help scientists and industry leaders tackle real-world challenges and make new discoveries. From life sciences and pharmaceutical research to food and contaminant analysis, environmental monitoring, forensics, and industrial quality control, our technologies and instruments provide the precision and reliability you need to make confident decisions.
Our innovative platforms - such as timsTOF, scimaX, neofleX, and DART-TQ - combined with advanced software like SCiLS™ Lab, MetaboScape®, and Biopharma Compass®, transform complex data into actionable insights. Breakthrough innovations like Trapped Ion Mobility (TIMS), Omnitrap®, and dual ionization GC-HRMS are redefining what’s possible in mass spectrometry.
Trusted by leading research institutes, universities, government agencies, and industrial partners worldwide, Bruker Daltonics is committed to driving scientific progress and delivering solutions that matter.
(For Research Use Only. Not for use in clinical diagnostic procedures).
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.