Solanum tuberosum (potato) is an essential crop for human nutrition and is known for its high cultural and agronomical significance. Resistance against biotic attacks is generally desired to sustain potato quality and reduce pesticide treatments.
To gain a better insight into the herbivory defense, an insect biting on the plants was simulated. Apart from the wildtype cultivar Désirée, the overexpression lines “AtACT-AtDTX18” were also analyzed. AtACT-AtDTX18 express a p-coumaroyl-CoA:agmatine N4-p-coumaroyltransferase from Arabidopsis thaliana and also an Arabidopsis thaliana MATE transporter to export a number of hydroxycinnamic acid amides. The AtACT-AtDTX18 overexpression plants are more resistant against Phytophthora infestans - the causal agent of late blight in potatoes and was responsible for Irish potato famine between 1845 and 1852.
Even today, it still leads to significant losses in crops. p-coumaroylagmatine - one of the hydroxycinnamic acid amides - inhibits the germination of spores in this agronomically significant oomycete.[1] A targeted analysis for hydroxycinnamic acid amides had earlier described the metabolic changes of the AtACT-AtDTX18 overexpression lines following Phytophthora infestans infection.[1] In this article, untargeted metabolite profiling was applied, predominantly focusing on the early metabolic responses to herbivory, which was replicated by damaging the leaves of the plant with tweezers. The article also addresses the question of whether the wounding responses in the overexpression lines having higher Phytophthora infestans resistance vary from those of the wildtype plants.
Several peaks of unknown metabolite structures are always revealed by metabolite profiling. This data set was thus used for a technique called “MS/MS similarity metabolite profiling.” Using MetaboScape, numerous compounds were annotated based on their MS/MS, which helped in providing a better understanding of potential potato defense metabolites.
Experiment
For the experiment, potato plants (Solanum tuberosum L. cv. Désirée and AtACT-AtDTX18 OE) were produced and grown for a period of four weeks in a phytochamber as illustrated in Dobritzsch et al.[1] Herbivore biting was simulated by wounding the leaves with tweezers and then about 500 mg of leaf material was obtained after eight hours. Using 200 µL 80% methanol, leaf metabolites were extracted two times. The following extracts were pooled, evaporated, and again dissolved in 30% methanol, followed by adjusting the final volume to the sampled material (final concentration of 50 mg fresh weight for each 100 µL extract). Using the devices and settings given in Table 1, extracts were subjected to UPLC-QTOF-MS/MS analysis. AutoMS/MS was carried out on three samples of each sample group. Then, recalibration of the MS data was carried out on lithium formate clusters toward the end of each run using DataAnalysis 4.4 (Bruker) for raw data, or entirely automatically by T-ReX 3D in MetaboScape 3.0 for metabolite profiling data.
Table 1. PLC-MS equipment and setup for metabolite profiling.
| Mass Spectrometry |
|
| Instrument |
MicrOTOF-Q II |
| Source |
Apollo II ESI source |
| Ionization |
ESI(+) |
| Scan Range |
m/z 50–1000 |
| Aquisition Rate |
3 Hz, autoMS/MS: 3 Hz |
| Calibration |
Internal calibration through automation at 12 min, Lithium Formate, HPC, Polarity: Positive |
| UPLC |
Waters Acquity |
| Column |
HSS T3 (100 x 1.0 mm) |
| Flow Rate |
0.15 ml/min |
| Mobile Phase |
A = water + 0.1% formic acid, B = acetonitrile + 0.1% formic |
| Gradient |
0 – 1 min 5% B
1 – 5 min to 75% B
5 – 10 min to 95% B
10 – 12 min 95% B
12 – 15 min 5% B |
| Data Processing |
MetaboScape 3.0 and DataAnalysis 4.4 |
Using the settings provided in Table 2, untargeted metabolite profiling was carried out in MetaboScape. AutoMS/MS was then matched on the generated bucket table. The four replicate groups - AtACT-AtDTX18 no wounding, AtACT-AtDTX18 wounding, S.t. cv. Désirée no wounding, and S.t. cv. Désirée wounding - were examined with 15–25 biological replicates each. Based on analytical standards and annotations in earlier experiments, “Analyte Lists” with over 2000 entries for targeted annotation were produced in MetaboScape. For analyte list matches, the confidence criteria were (annotation quality: very good/acceptable): m/z: 2/20 ppm; retention time: 0.1/0.2 minutes; MS/MS score: 900/800; and mSigma: 20/60.
Table 2. MetaboScape 3.0 processing parameters.
| . |
. |
| Intensity threshold [counts] |
1500 |
| Minimum peak length [spectra] |
10 |
| Minimum peak length (recursive) [spectra] |
8 |
| Minimum # Features for Extraction |
31 |
| Presence of features in minimum # of analyses |
31 |
| Lock mass calibration |
false |
| Mass calibration |
true |
| Calib. RT start |
723 |
| Calib. RT end |
732 |
| Filter by sample group |
S.t. AtACT-AtDTX18 overexpression vs S.t. cv. Désirée |
| Minimal value of features in sample group (type) |
PERCENTAGE |
| Minimal value of features in sample group |
75 |
| Area |
true |
| Primary Ion |
[M+H]+ |
| Seed Ions |
[M+Na]+ |
| EIC correlation |
0.9 |
| Mass range: Start [m/z] |
50 |
| Mass range: End [m/z] |
1000 |
| Retention time range: Start [min] |
0.6 |
| Retention time range: End [min] |
10 |
| Perform MS/MS import |
false |
| Group by collision energy |
false |
| MS/MS import method |
maxsum |
Pathway maps were subsequently drawn in PathVisio 3.3.0[2] depending on the information from KEGG pathways[3] (phenylpropanoid biosynthesis/arginine and proline metabolism) with Arabidopsis thaliana and Solanum tuberosum as reference organisms.
Results
MetaboScape - Metabolite Profiling of Unwounded and Wounded Solanum tuberosum Plants
Using the chosen settings for peak picking as shown in Table 2, the higher abundant leaf metabolites were given focus.
A bucket table of 440 regions with complete extracted features was generated by the MetaboScape T-ReX 3D peak picking algorithm. For a majority of these, the MS/MS were assigned from autoMS/MS. The analyte lists offered annotations for 96 of the 440 metabolites that corresponded with the criteria of accurate retention time, isotopic pattern, and m/z. A quick and reliable assessment of the identification confidence was enabled by the intuitive graphical annotation quality symbols.
Therefore, the simplified data processing in Metaboscape enabled identification for about 25% of the features. This underscores the fact that there are many specific metabolites in potato yet to be covered by the utilized Analyte List.
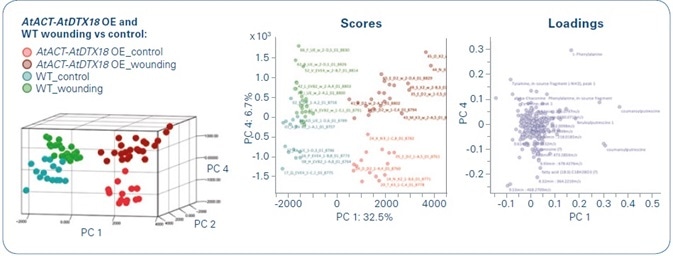
Figure 1. Principal component analysis of wounded and unwounded potato wildtype (WT) and AtACT-AtDTX18 overexpression (OE) lines.
The first impression of sample similarity and treatment-based metabolic differences was offered by a principal component analysis, or PCA. PC 1 showed a clear difference between the wildtype and the overexpression lines (OE), while the groups of wounded versus unwounded plants were separated in PC 4 (Figure 1). The PCA’s loading values showed coumaroylputrescine, coumaroylagmatine, caffeoylputrescine, and feruloylputrescine as the most pertinent PC 1 metabolites. The improvement of these hydroxycinnamic acid amides (HCAAs) in the OE lines was previously described in a targeted analysis by the group.[1] This analysis provided an understanding of the high MS/MS similarity of HCAAs that have indicative main fragments and systematic neutral losses. This formed the basis for a new data analysis, which was called “unsupervised MS/MS similarity metabolite profiling.”
Annotation of Metabolites by Unsupervised MS/MS Similarity Metabolite Profiling
For a majority of non-identified compounds, MS/MS information was available and thus a high degree of structural data in general. Metabolites that are structurally similar usually exhibit analogous MS/MS. Therefore, the comparison of MS/MS from unknowns to annotated features offers the potential of unknown identification. Yet, one by one comparison of a countless number of MS/MS is difficult to handle manually.
MetaboScape provides the tools needed to analyze MS/MS data, with focus given to compounds that are structurally similar. The following strategy was applied: The key PC1 features were chosen for “MS/MS bucket matches” (see Figure 2).
![MS/MS similarity metabolite profiling. The MS/MS of p-coumaroylputrescine (cis/trans-isomer 2) was selected for “MS/MS bucket matches” (bucket highlighted in blue in the center). Hydroxycinnamic acid amides (HCAAs) are stereoisomers and occur as two LC-separated peaks, which are termed as “cis/trans 1 or 2.” Of the 11 matching MS/MS, 6 compounds were identified as HCAAs [compounds 1, 2, [2 isomers] and [2 isomers]]. Compound [2 isomers] could be described by in-depth MS/MS analysis as coumaroylagmatine with a modified agmatine residue; the compounds in box were flagged as tentative HCAA-like compounds.](https://www.news-medical.net/image-handler/picture/2019/3/Job_6039_Art1_Pic2.jpg)
Figure 2. MS/MS similarity metabolite profiling. The MS/MS of p-coumaroylputrescine (cis/trans-isomer 2) was selected for “MS/MS bucket matches” (bucket highlighted in blue in the center). Hydroxycinnamic acid amides (HCAAs) are stereoisomers and occur as two LC-separated peaks, which are termed as “cis/trans 1 or 2.” Of the 11 matching MS/MS, 6 compounds were identified as HCAAs [ 1, 2, [2 isomers]
1, 2, [2 isomers]  and [2 isomers]]. Compound compounds
and [2 isomers]]. Compound compounds  [2 isomers] could be described by in-depth MS/MS analysis as coumaroylagmatine with a modified agmatine residue; the compounds in box were flagged as tentative HCAA-like compounds.
[2 isomers] could be described by in-depth MS/MS analysis as coumaroylagmatine with a modified agmatine residue; the compounds in box were flagged as tentative HCAA-like compounds.
This tool compares the MS/MS of the chosen compound with all MS/MS in the bucket table. This rapidly showed various features that had potential structural similarity to the specified hydroxycinnamic acid target. Moreover, among the MS/MS matching buckets, there were compounds of already annotated HCAAs (for example, p-coumaroylagmatine, p-coumaroylspermidine), which verified the method. This also indicated several unknowns, earlier unidentified compounds that had MS/MS similarity. With the help of MetaboScape, compounds of interest can be flagged, for instance, for in-depth analyses at a later stage.
This flagging was applied for emphasizing a number of HCAA-like compounds, as shown in Figure 2. After a more comprehensive analysis of the MS/MS, numerous structures of the HCAA compound family that have not been described so far were annotated. This approach was applied to other features (caffeoyl- and feruloyl-amides) and a total of 24 HCAAs was observed. As a result, the MetaboScape software made it possible to extract several compounds with just a single click that revealed structural similarity. In total, 113 metabolites could be annotated, as illustrated in Figure 3.
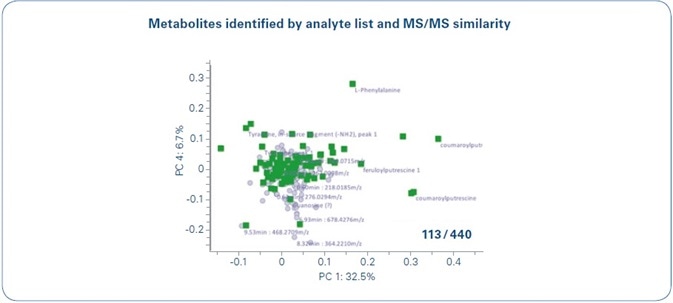
Figure 3. Metabolite identification and annotation in MetaboScape. 13 out of 440 buckets were annotated in this work. 96 buckets were assigned based on Analyte Lists and 17 could be assigned as HCCAAs by MS/MS similarity analyses. To visualize that these 113 metabolites correspond to the main PCA loadings relevant in this study (see Figure 1), all annotated loadings are highlighted in green in the PC1 vs. PC 4 loadings plot.
Here, it must be highlighted that almost all HCAAs were of higher abundance in the overexpression lines. The tool “Pathway Mapping” in the MetaboScape software was used to view this usual enhancement and to underscore common biosynthetic origins. Many new and additional metabolites - annotated during the MS/MS similarity method detailed above - helped in extending a pathway map. In addition, it was possible to incorporate the reactions that were catalyzed by the overexpressed AtACT gene product based on the data obtained from KEGG.[3] In MetaboScape, the fold change mapping (see Figure 4) indicated two major findings:
- The AtACT overexpression influenced the abundance of its reported direct metabolic products, and almost all HCAAs were changed and increased in the OE line
- Feruloylagmatine, albeit known to be a catalytic product of the AtACT enzyme, was decreased after the enzyme was overexpressed
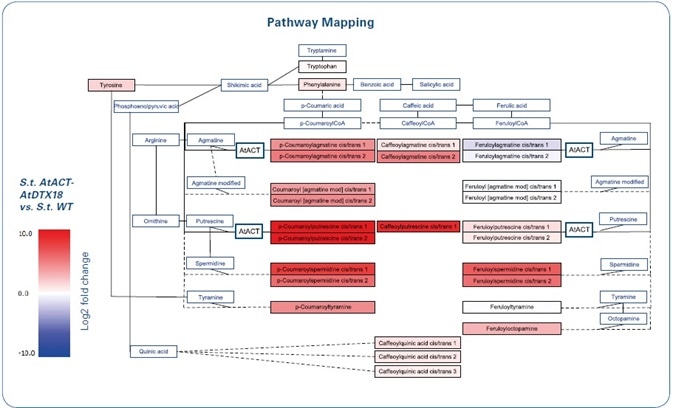
Figure 4. Extended biosynthetic pathway of hydroxycinnamic acid amide biosynthesis in potato (Solanum tuberosum (S.t.)), that was genetically modified by the introduction of the Arabidopsis genes AtACT and AtDTX18. AtACT catalyzes the biosynthesis of p-coumaroylagmatine and p-coumaroylputrescine as well as feruloylagmatine and feruloylputrescine in Arabidopsis (KEGG pathways). The HCAA pathway was adapted from KEGG to include all detected HCAA structures. Colors indicate the fold change of the overexpression lines versus the wildtype (without wounding).
Therefore, there may be instant downstream degradation of feruloylagmatine, the Arabidopsis enzyme may have distinct substrate specificity in the metabolic context of potato, or precursors might be restricted. This underscores the fact that the MetaboScape workflow offered many findings for more interesting follow-up experiments.
Wound-Induced Metabolites in Potato
The separate comparison of control and treatment in both genotypes - wildtype and overexpression lines - clearly revealed the metabolic changes in response to wounding. Common wound-induced features were revealed by the t-test results (see Figure 5).
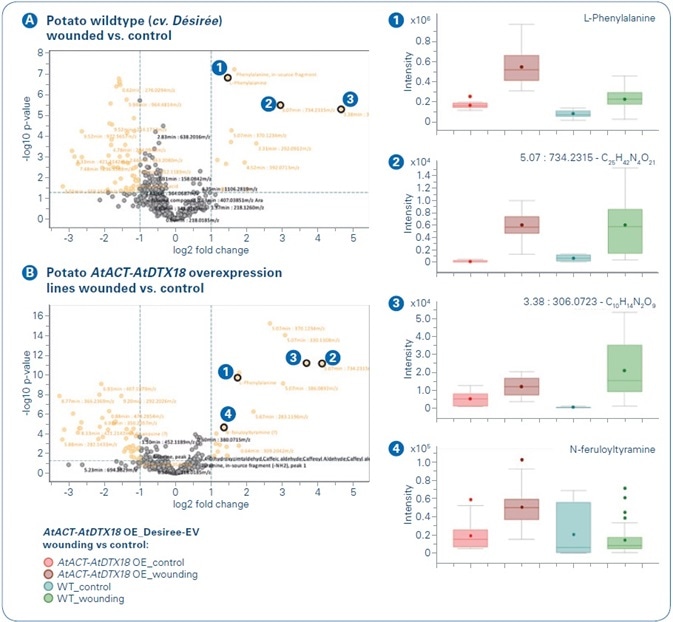
Figure 5. Wound-responsive buckets in potato (Solanum tuberosum cv. Désirée) wildtype  and AtACT-AtDTX18 overexpression lines
and AtACT-AtDTX18 overexpression lines  : MetaboScape generated volcano plots based on t-test results ( and ) and boxplots (
: MetaboScape generated volcano plots based on t-test results ( and ) and boxplots ( ) are shown.
) are shown.
Phenylalanine was considerably increased after wounding in both OE and WT plants (see Figure 5, compound 1), which too was implied by the PCA with phenylalanine as PC 4 loading value (see Figure 1). Furthermore, the molecular formulae of other regular wound-induced features were tentatively assigned using Smart Formula (see Figure 5, compounds 2 and 3). Several metabolites were wound-induced in the AtACT-AtDTX18 overexpression lines, but not considerably in the wildtype plants eight hours post wounding. Feruloyltyramine was the most fascinating feature (see Figure 5, compound 4).
A compound of the HCAA metabolite clade, feruloyltyramine was already elucidated as a Phytophthora infestans-responsive metabolite in potato by Keller and team in 1996[4] and as a wound-responsive compound in potato tubers by Negrel and team in 1993.[5] By contrast to both teams, no major increases of feruloyltyramine were observed in the wounded WT plants, which may be explained by a previous sampling time point (8 hours rather than 6 days (Negrel et al.) or 24 and 48 hours (Keller et al.)) or differential reactions in response to wounding and towards the oomycete elicitor.
Conclusion
- With the help of the information obtained with Bruker LC-ESI-Q-TOF MS and MS/MS instrumentation and techniques, the MetaboScape software was able to offer an efficient platform for the simple and easy linkage of metabolite abundances and structural similarities. Numerous statistical tools (t-test, PCA) were used in MetaboScape to concentrate on pertinent features. The unsupervised MS/MS similarity metabolite profiling method made it possible to rapidly annotate 17 metabolites, which were earlier unknown in the analyses.
- The combination of regular abundance patterns and common MS/MS patterns indicated an overflow of the hydroxycinnamic acid amide biosynthesis in the AtACT-AtDTX18 overexpression lines. This is considered to be perfect according to the function of the AtACT overexpressed enzyme that plays a role in the biosynthesis of four HCAAs. In addition, the increase in many other HCAAs could be the plant’s metabolic side effect to offset this overflow and to bypass deadly effects for the cell. The MetaboScape software proved to be highly valuable for convenient data processing, enabling the annotation of various previously unknown compounds as well as an intuitive visualization of metabolic changes.
References
[1] Dobritzsch M, Lubken T, Eschen-Lippold L, Gorzolka K, Blum E, Matern A, et al. MATE Transporter- Dependent Export of Hydroxycinnamic Acid Amides. Plant Cell. 2016;28(2):583-96.
[2] Kutmon M, van Iersel MP, Bohler A, Kelder T, Nunes N, Pico AR, et al. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLOS Computational Biology. 2015;11(2):e1004085.
[3] Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research. 2017;45(D1):D353-D61.
[4] Keller H, Hohlfeld H, Wray V, Hahlbrock K, Scheel D, Strack D. Changes in the accumulation of soluble and cell wall-bound phenolics in elicitor-treated cell suspension cultures and fungus-infected leaves of Solanum tuberosum. Phytochemistry. 1996;42(2):389-96.
[5] Negrel J, Javelle F, Paynot M. Wound-Induced Tyramine Hydroxycinnamoyl Transferase in Potato (Solanum tuberosum) Tuber discs. Journal of Plant Physiology. 1993;142(5):518-24.
About Bruker Daltonics
Empowering Science – Improving Life
Bruker Daltonics delivers cutting-edge mass spectrometry solutions and workflows that help scientists and industry leaders tackle real-world challenges and make new discoveries. From life sciences and pharmaceutical research to food and contaminant analysis, environmental monitoring, forensics, and industrial quality control, our technologies and instruments provide the precision and reliability you need to make confident decisions.
Our innovative platforms - such as timsTOF, scimaX, neofleX, and DART-TQ - combined with advanced software like SCiLS™ Lab, MetaboScape®, and Biopharma Compass®, transform complex data into actionable insights. Breakthrough innovations like Trapped Ion Mobility (TIMS), Omnitrap®, and dual ionization GC-HRMS are redefining what’s possible in mass spectrometry.
Trusted by leading research institutes, universities, government agencies, and industrial partners worldwide, Bruker Daltonics is committed to driving scientific progress and delivering solutions that matter.
(For Research Use Only. Not for use in clinical diagnostic procedures).
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.