For glycopeptide characterization, mass spectrometry has arisen as the go-to methodology. As well as the instrumental arrangement, a suitable software solution is essential to enable glycopeptide identification and cut laborious processing and de novo analysis time.
GlycoQuest (search algorithm in ProteinScape) is Bruker’s proven solution and delivers dependable results with regards to glycopeptide identification.
This article discusses a novel, enhanced classification workflow for glycopeptides evaluated with the Glycopeptide Instant Expertise™ acquisition method (QTof series).
It is possible to achieve glycopeptide fragmentation under standard collision induced dissociation (CID) conditions, in which the glycosidic bonds between carbohydrate bonds are favourably cleaved.
Nevertheless, this seldom yields peptide b- and y-type ions (cleavage of peptide bond), which, in many instances, renders peptide identification impossible. Conversely, higher-energy CID (HCD) mainly leads to b/y-type peptide ions, as well as glycan oxonium ions and fewer Y-type ions from fragmentation of the glycosidic linkage.
Hinneburg et al.1,2 discussed a new glycopeptide instant expertise method which makes use of multiple collision energies to produce glycan fragments and peptide fragments inside the same MS/MS spectrum.
With the combination of the process of fragmentation and the new classification workflow in ProteinScape, it is possible to determine glycan composition and peptide sequence simultaneously and reduce the number of false or wrongly classified spectra.
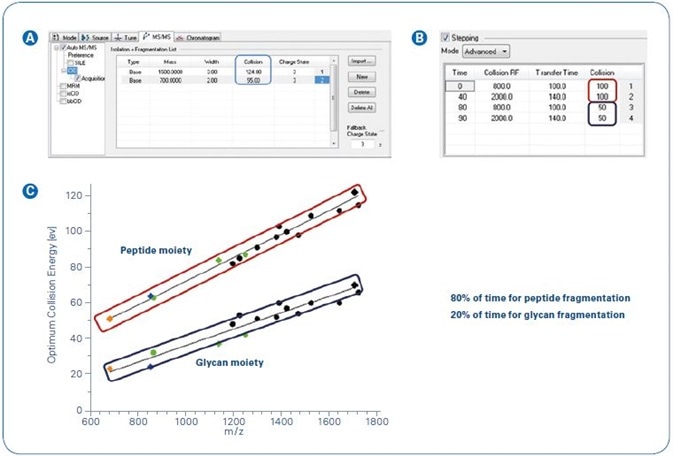
Figure 1. Collision energy stepping implemented in the Glycopeptide Instant Expertise™ method.
Experimental
Sample
Any variety of glycopeptide sample can be employed in this workflow – low complex and high complex, with and without glycopeptide enrichment.
Instrumentation
The measurement can be carried out with either a direct infusion setup or LC separation.
Mass Spectrometry Instrument
Compact, Impact II, MaXis II Method: Glycopeptide Instant Expertise™
Data Processing
Compound classification and ensuing glycopeptide identification is executed with ProteinScape 4.0 and the embedded search algorithm GlycoQuest.
Results and Discussion
Until recently, classification of N-glycopeptide spectra in ProteinScape (detection of glycopeptide spectra and calculation of glycan and peptide moiety mass) has centered on glycan fragment distances and oxonium ion detection when using typical CID conditions on ion trap or qTof instruments (see application note LCMS-66)4.
While [peptide + GlcNAc + H]+ is always the final fragment in the glycan cleavage chain of ion trap spectra (resonant fragmentation), qTof fragmentation generally produces spectra with either [peptide + GlcNAc + H]+ or [peptide + H]+ as the final fragment in the chain, dependent on the size of the peptide and glycan moiety, and charge distribution across the molecule.
Despite the fact that the classification workflow in ProteinScape has an explicit parameter to address this issue, the peptide mass of low abundant N-glycopeptides can be miscalculated at times, which can result in no or incorrect identifications.
To find a way around this issue, deeper investigation of the fragmentation of N-glycopeptides was carried out using Bruker oTOF instruments.
Hinneburg et al.1,2 employed synthetic, and therefore well-defined, N-glycopeptides for the optimization of CID energy parameters, so as to attain the greatest possible quantities of data on both the glycan and peptide moiety within just one tandem MS experiment.
Figure 1C shows the ideal collision energies. The application of these results into a consistent acquisition method was managed through the use of a timed stepping of collision energies and transfer settings (Figure 1A and B).
In the case of N-glycopeptide fragmentation, the higher collision energies, which are required for peptide moiety fragmentation, also bring about a characteristic fragmentation pattern, which has earlier been described for MALDI fragmentation3.
In these settings, (0,2)X-ring fragmentation of the innermost N-acetylglucosamine of the chitobiose core (+83 Da), and a loss of the complete N-glycan moiety by cleavage of both the N-glycosidic bond and the side-chain amide group of the N-glycosylated asparagine, produces a characteristic peak doublet with a mass difference of 17 Da.
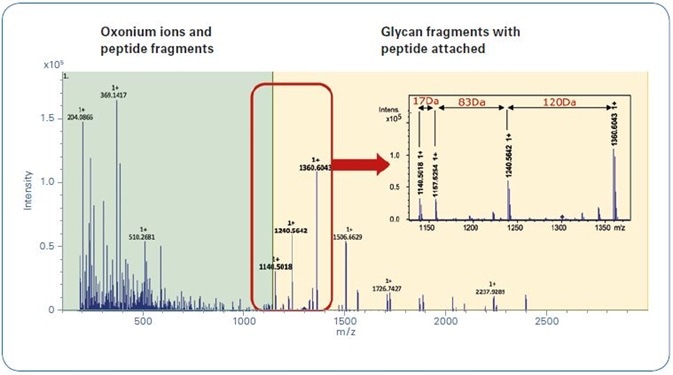
Figure 2. Typical glycopeptide CID spectrum achieved by using the Glycopeptide Instant Expertise™ method.
Figure 2 shows a characteristic CID N-glycopeptide CID spectrum, measured with the Glycopeptide Instant Expertise™ acquisition method. The zoom draws attention to the fragmentation pattern mentioned earlier.
As ProteinScape also allows for the classification of MALDI glycopeptide fragment spectra, the software comes with a built-in classification workflow based on the characteristic fragments seen in Figure 2 and outlined above (-17 Da, [Peptide + H]+, +83 Da, +120 Da).
Additionally, this workflow can be used for qTof fragmentation, where it adds a further typical trait of N-Glycopeptide spectra that improves the reliability of automated glycopeptide spectra classification (see Figure 3). Figure 4 offers an example for a classified (new workflow) and identified (by GlycoQuest and Mascot) N-glycopeptide spectrum.
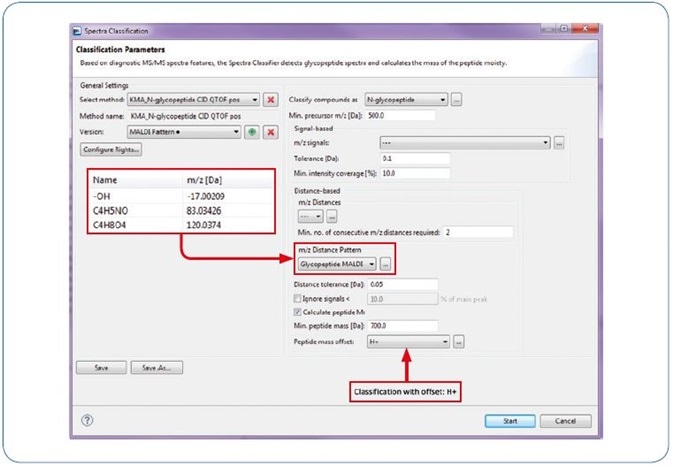
Figure 3. ProteinScape: Classification with “MALDI Pattern”.
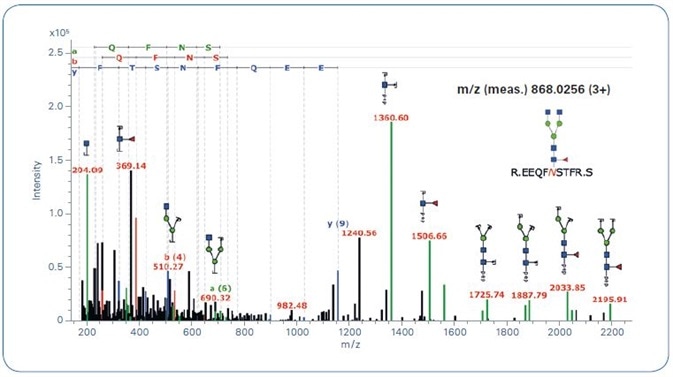
Figure 4. ProteinScape: Annotated glycopeptide CID spectrum.
Conclusions
N-glycopeptide analysis can be enhanced through the adaptation of CID fragmentation parameters, as described by Hinneburg et al.1,2, combined with a targeted classification workflow in ProteinScape.
Thanks to the addition of an explicit diagnostic fragmentation pattern, the quantity of wrongly calculated peptide masses is radically reduced, resulting in extremely confident N-glycopeptide identification results.
References
- Hinneburg H, Stavenhagen K, Schweiger-Hufnagel U, Pengelley S, Jabs W, Seeberger PH, Silva DV, Wuhrer M, Kolarich D. The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. J Am Soc Mass Spectrom. 2016 Mar;27(3):507-19.
- Poster Note PN-03: Using multiple collision energies to improve N- and O-glycopeptide identification by LC-MS/MS. Hinneburg H, Kolarich D, Schweiger-Hufnagel U, Pengelley S; Bruker Daltonik GmbH, Bremen, Germany.
- Wuhrer M, Hokke CH, Deelder AM. Glycopeptide analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry reveals novel features of horseradish peroxidase glycosylation. Rapid Commun Mass Spectrom. 2004;18(15):1741-8.
- Application Note # LCMS-66: Straightforward N-glycopeptide analysis combining fast ion trap data acquisition with new ProteinScape functionalities. Neue K, Kiehne A, Meyer M, Macht M, Schweiger-Hufnagel U, Resemann A, Bruker Daltonik GmbH, Bremen, Germany
About Bruker Daltonics
Empowering Science – Improving Life
Bruker Daltonics delivers cutting-edge mass spectrometry solutions and workflows that help scientists and industry leaders tackle real-world challenges and make new discoveries. From life sciences and pharmaceutical research to food and contaminant analysis, environmental monitoring, forensics, and industrial quality control, our technologies and instruments provide the precision and reliability you need to make confident decisions.
Our innovative platforms - such as timsTOF, scimaX, neofleX, and DART-TQ - combined with advanced software like SCiLS™ Lab, MetaboScape®, and Biopharma Compass®, transform complex data into actionable insights. Breakthrough innovations like Trapped Ion Mobility (TIMS), Omnitrap®, and dual ionization GC-HRMS are redefining what’s possible in mass spectrometry.
Trusted by leading research institutes, universities, government agencies, and industrial partners worldwide, Bruker Daltonics is committed to driving scientific progress and delivering solutions that matter.
(For Research Use Only. Not for use in clinical diagnostic procedures).
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.