This article takes a closer look at the multi-legislative compliance process for medical devices and in-vitro diagnostic medical devices (IVDs). Expanding on what was discussed in the first installment about product safety legislation and horizontal legislation, this article will focus on what manufacturers must do to comply with sectorial, horizontal, and national regulations.
Applicable harmonization (sectorial) legislation
Since the 2017 publication of the medical device regulation (MDR) and the IVD regulation (IVDR), EU regulations for medical devices and IVDs have undergone a profound change. Manufacturers are now well-experienced in the application of both regulations and their alignment with the new legislative framework and market surveillance regulations.1,3
Concepts, including economic operators, market surveillance, customs controls, and enforcement, are significantly reinforced in the sectorial legislation and far better comprehended by industry.
Beyond the primary legislation, MDR and IVDR also introduced multiple pieces of secondary legislation (implementing acts and delegated acts) that manufacturers should be aware of. Examples include:
- Regulation (EU) 2021/2226 on electronic instructions for medical device use
- Regulation (EU) 2022/2347 on the reclassification of groups of certain active products without an intended medical purpose
- Regulation (EU) 2023/2197 on the assignment of Unique Device Identifiers for contact lenses
These implementing acts and delegated acts provide procedural and technical details to support MDR and IVDR adoption and must be applied during conformity assessments whenever relevant.
National legislation
Although the MDR and IVDR apply directly to all EU Member States, specific elements necessitate or permit national implementation or additional national rules. National legislation may include provisions for:
- Clinical investigations
- Market Surveillance and Enforcement/fines
- Registration of Economic Operators
- Language and Labeling Requirements
Manufacturers should ensure they understand applicable national legislation and integrate it into their market access approaches.
Relevant horizontal legislation
However, manufacturers may underestimate the underlying complexity of applying EU legislation. CE marking a medical device or IVD commonly requires a hybrid approach, taking into account both the sector-specific harmonization legislation and other horizontal legislation. An analogy can be made with standards where both sectorial (product standards) and horizontal standards (e.g. ISO 14971) may apply.
Generally, CE marking is mandatory for products that address health, safety, environmental, and consumer protection requirements. Currently, the EU has 30 different types of harmonization legislation (sectorial) aligned with the novel legislative framework approach.
CE marking is required for products covered by harmonization legislation that explicitly mandate it. Several noteworthy examples with the potential to apply to medical devices include:
- RoHS – Directive 2011/65/EU
- Electromagnetic Compatibility – Directive 2014/30/EU
- Radio equipment – Directive 2014/53/EU
- Low Voltage – Directive 2014/35/EU
- Batteries – Regulation (EU) 2023/1542
- Machinery – Regulation (EU) 2023/1230
- Artificial Intelligence Act – Regulation (EU) 2024/1689
- Packaging and Packaging Waste – Regulation (EU) 2025/40
Other EU regulations, while not harmonization legislation themselves, interact with it. In other words, they do not require a CE mark in and of themselves in isolation. These regulations are best understood as horizontal legislation underpinning and complementing sectorial legislation.
Horizontal rules are cross-sectorial regulations or guidelines that apply across multiple sectors or policy areas, instead of being restricted to a certain industry or product type. They are designed to maintain consistency, coherence, and fairness in the application of EU legislation. Some key examples with the potential to apply to medical devices include:
- REACH Regulation – Regulation (EC) No 1907/2006
- CLP Regulation (EC) No 1272/2008
- Product Liability Directive – Directive 85/374/EEC / Directive (EU) 2024/2853
- General Data Protection Regulation (GDPR) – Regulation (EU) 2016/679
In most cases, CE marking a medical device cannot be achieved solely based on compliance with the medical device or IVD sectorial legislation. In addition to the MDR or the IVDR, multiple other sectorial regulations and horizontal regulations may apply.
When CE marking a device, all relevant EU legislation should be applied and captured in a single declaration of conformity. This makes CE marking more than just an exercise in complying with the MDR or IVDR.
Medical device software may be an exception, as it can be delivered and consumed electronically and is essentially a digital asset. Therefore, other regulations are less likely to apply compared to physical devices.
However, as far as defective products and financial coverage are concerned, even software is subject to the product liability directive. In addition, other EU rules related to artificial intelligence and cybersecurity may apply to software.
Implications for QMS design
Manufacturers must proactively ensure that their quality management systems are designed to meet all relevant regulatory requirements for their products. Historically, manufacturers usually designed their QMSs to specifically comply with the requirements of ISO 13485, MDSAP, and the medical device regulations in both the EU and USA.
However, to address all aspects of applicable regulations, it is increasingly necessary to design the QMS more broadly. For instance, understanding whether the QMS requires the consideration of other regulations as sources of design input or labeling is essential.
Example: Active implantable device
Consider a manufacturer of a fictitious active implantable device. The device contains a battery, electrical circuits/components, software/firmware, and can communicate with a controller or programmer through radio frequency communications.
In this example, the primary legislation for the device’s CE marking is the medical device sectorial legislation, and in this case, a Notified Body designated under the MDR will perform the conformity assessment.
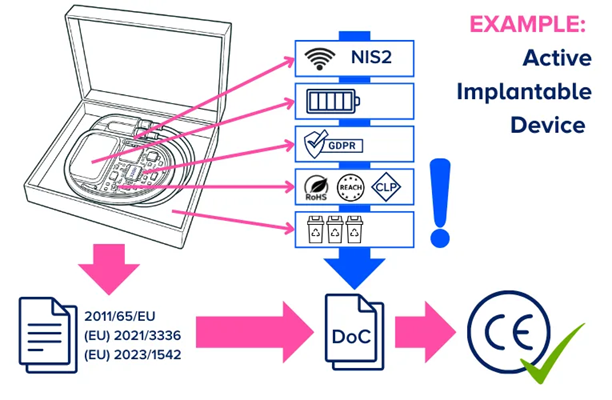
Image Credit: RQM+
However, the device will also fall under the RoHS directives (for the electrical circuits/components materials), the radio equipment directive (for communications), the NIS 2 Directive (for cybersecurity), the batteries regulation, and the packaging regulation.
Moreover, the materials will require assessment in accordance with the REACH and CLP regulations. GDPR applies if the device collects, stores, transmits, or otherwise processes personal data.
In this case, CE marking can only be achieved once all applicable requirements from these directives and regulations have been met, and a single declaration of conformity has been drawn up. This could require further technical documentation and objective evidence to demonstrate compliance.
Although the Notified Body mainly focuses on reviewing the objective evidence related to MDR compliance, they can equally inquire about the evidence to support other regulations.
New and changing regulations
There remains an ever-growing stream of new and evolving EU regulations that may impact medical devices. These regulations often lie outside the main competencies of conventional medical device companies and their R&D, QA, and RA departments.
Companies that are able to adapt, upskill, and modernize will certainly gain a significant advantage in the future for compliance and market access. The key enabler is constructing a management system grounded in all applicable regulations, not solely the narrow sectorial legislation for medical devices, which has traditionally been the case.
Manufacturers capable of developing a comprehensive understanding of EU regulations and integrating it into their regulatory intelligence process will be well-positioned for future success.
Summary
CE marking a medical device or IVD requires a comprehensive analysis of all applicable harmonization legislation, horizontal regulations, and national legislation of member states. CE marking based on medical device regulation or IVD regulation alone is rarely adequate for achieving a fully compliant and marketable device. Manufacturers must develop extensive and comprehensive market access approaches to address all applicable requirements.
Within the broader EU legal framework, the MDR and IVDR present multiple complexities and challenges that extend beyond the regulations themselves. These challenges originate from MDR and IVDR interactions with other EU laws, institutions, and market dynamics. This multi-layered compliance increases complexity, particularly for SMEs lacking regulatory resources.
Related next steps
Manufacturers facing challenges with the EU regulatory framework and compliance with horizontal EU regulations can rely on RQM+ Corp experts who specialize in developing comprehensive regulatory strategies designed to ensure that medical devices and IVDs meet all relevant EU requirements.
Acknowledgments
Produced from materials originally authored by Chris A. Parr, Principal, CRO at RQM+ Corp.
References and further reading:
- European Union. (2021). Regulation - 765/2008 - EN - EUR-Lex. (online) Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32008R0765.
- European Union. (2024). Decision - 2008/768 - EN - EUR-Lex. (online) Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32008D0768.
- European Union. (2019). EUR-Lex - 32019R1020 - EN - EUR-Lex. (online] Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32019R1020.
About RQM+
Two industry leaders - R&Q and Maetrics - merged in 2020 to become RQM+, the largest medical device and diagnostics-focused full-service regulatory and quality consulting firm in the world. The integration of our leadership teams and the extensive skills and experience of our collective resources have created a highly agile and scalable global organization.
With offices in the United States, United Kingdom, and Switzerland, our large global footprint allows us to meet the regulatory and quality support needs of international clients of all sizes.
Order an Audit
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.