If DNA is the fundamental code of the genome, RNA is the message. Genes are made active through the process of transcribing the code into RNA, and then translating RNA into protein. This is known as the central dogma of molecular biology.
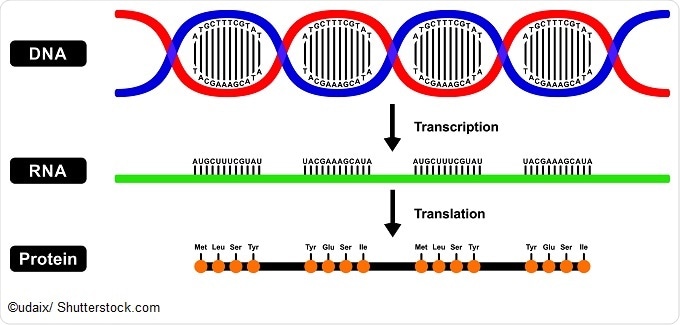
Recently, it’s become apparent that RNA has many other roles to play than as a messenger for the DNA code. RNA can have enzymatic activity, regulate gene expression, and transmit signals. RNA is also an important part of the ribosome, a cellular machine for assembling proteins.
The sequence of all RNAs in an organism is known as the transcriptome. Just as sequencing the genome has been important for understanding the structure and function of genes, sequencing the transcriptome can reveal the diverse functions and interactions of all of the RNA molecules in an organism.
But unlike the genome, the transcriptome varies depending on the stage of life of the organism and its physiological condition. The transcriptome is thus proportionately larger and more complex than the genome.
The use of next generation sequencing
Next generation sequencing technology has been used to develop RNA-Seq, a high throughput, deep sequencing approach for transcriptomes. Previous RNA sequencing methods have been based on high-density microarrays or genomic tiling. These methods are limited in that they rely on existing knowledge of the sequence and can be complicated to analyze.
RNA-seq reads the sequence directly. To begin, total or fractionated RNA is converted to DNA through reverse transcription, a process of writing the RNA code back into DNA. This results in a complementary (cDNA) library. The cDNA is then sequenced using next generation sequencing technology--usually Illumina, SOLiD, or Roche 454. The resulting sequences are then either assembled from scratch or aligned with a known sequence to create a transcription map.
Data Analysis
RNA-seq generates huge quantities of data, requiring massive computing resources to process and analyze. RNA sequencing analysis can also be complicated by splicing, a process in which precursor messenger RNA is edited to remove introns. Certain genes can also be subject to alternative splicing, the process whereby a single RNA can be translated into different proteins through multiple splicing sites, and trans splicing, where exons from two different RNA transcripts are spliced end to end.
Depth
Unlike DNA, RNA transcripts vary in abundance. Depth in sequencing is required to sequence all transcripts, including those that are rare. Depth is described as the number of unique reads, and typically varies from a low of a few million to over 100 million. The larger the genome, the more sequencing depth is required.
Applications
RNA-seq has been used for a broad range of studies. For example, it was used to identify hundreds of differentially expressed genes in psoriasis in 2009, opening up new possibilities for treatment of the disease. A 2012 study revealed unexpected complexity in the human transcriptome when RNA-seq was paired with genomic tiling arrays.
That complexity was beyond expectations and still challenges the boundaries of existing sequencing technology. RNA-seq has also been used in plant science, for example to characterize the gene expression profile of maturing citrus fruit.
Sources
- http://www.nature.com/nrg/journal/v10/n1/full/nrg2484.html
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3381505/
http://www.nature.com/nbt/journal/v30/n1/full/nbt.2024.html
- https://peerj.com/articles/3343/
Further Reading
Last Updated: Feb 26, 2019
 Gut microbes may reveal who is at risk of type 2 diabetes years before diagnosis
Gut microbes may reveal who is at risk of type 2 diabetes years before diagnosis