Lysosomes are subcellular organelles that are present in most cells, with the major exception of red blood cells.
They have the clearing function in the body, functioning like a garbage disposal and include enzymes called hydrolases which digest what is present in the cells, including proteins, lipids, sugars, and nucleic acid.
Where it becomes a problem is if these lysosomes or enzymes within the lysosomes, don’t work correctly, problems tend to arise.
The proteins, acids or sugars are generated and build up within the cell, as they cannot be broken down, and that can ultimately lead to cellular tissue and organ damage.

Which of these functions is defective in Fabry disease and what effect does this have?
In Fabry disease, a protein called alpha-galactosidase A is deficient. The gene that codes for this enzyme is defective, meaning that the body either creates no enzyme at all, or develops an enzyme which doesn’t function properly.
Due to the lack of alpha-galactosidase A, a fatty substance called globotriaosylceramide (Gb3) builds up in the lysosomes.
Because lysosomes are present in almost any tissue or cell in the body, different complications and clinical symptoms start to arise.
The lack of the enzyme can affect the central nervous system, ears, eyes, gastrointestinal tract, heart, kidneys, the skin and most importantly the cardiovascular system.
What is interesting, and scientifically and clinically challenging, is the fact that Fabry disease, by definition, is an X-linked disease; therefore, it was once thought that women were just carriers and only men were clinically affected.
However, there is a process called lyonization, which basically means there is an X chromosome inactivation of a gene. Women will have this gene twice, because they have two X chromosomes.
In women, one copy of the X chromosome is inactivated which means that for women, as far as Fabry disease is concerned, the inactivation that takes place is highly different across tissue: women may have one tissue or organ where the gene is inactivated and another one where it isn’t, which leads to highly variable clinical symptoms.
For example, the only clinical symptom you might observe is that a woman has a stroke as a first symptom of Fabry disease at a young age.

What are the main signs and symptoms of Fabry disease?
Fabry disease is a progressive disorder and the number of organs that are affected can vary and develop over time, as can the severity of the symptoms. As you would expect, they increase with increasing age.
Signs and symptoms are different in each patient and that’s one of the fundamental challenges in diagnosing Fabry disease early.
The first signs of Fabry disease typically start in childhood: in boys, between the age of three and ten years, and girls are generally affected a couple of years later, on average ten years or so later.
60-80% of the classically affected patients report pain as their first symptom. This can be very tough for children.
Other symptoms that appear are skin lesions; healing impairment; sweating abnormalities; physical performance issues, the sense of feeling tired and fatigued; and gastrointestinal symptoms.
As the disease progresses, patients can develop renal symptoms and declining renal function, as well as heart problems such as left ventricular hypertrophy, arrhythmias and stroke at a very early age.
The major morbidity, and what’s really dangerous in Fabry disease, is the risk of end-stage renal disease and life-threatening cardiovascular or cerebrovascular disease. These are the morbidities that shorten the life expectancy of Fabry patients.
While some data, in the past, indicated that end-stage renal disease was the major cause of death, nowadays it seems that cardiovascular disease and cardiac morbidity and mortality in particular, is what impacts a patient’s life expectancy to a larger extend then renal problems.

How is Fabry disease currently diagnosed?
Diagnosing Fabry disease is the big challenge from a clinical perspective. Rare diseases in general are challenging to diagnose because physicians are trained that frequent things are frequent and rarely see patients with this disease. Therefore, we’re not very good at diagnosing rare diseases.
For example, during my clinical training and time spent working in a hospital, I never saw a Fabry patient myself. The first time I actually met a Fabry patient was in my role at Shire, so there’s a fundamental challenge for diagnosing rare diseases because of the lack of exposure.
The classical symptom that used to lead to diagnosis was stroke, however, these days patients are typically diagnosed because they start to develop kidney problems, i.e. microalbuminuria or a decline of glomerular filtration. Patients may also present with clinical heart symptoms.
Many patients are also found in dialysis units, where it is discovered that the reason for the end-stage renal failure is in fact Fabry disease.
The other way that patients can be diagnosed is linked to the genetic nature of the disease. If you have one patient in a family who has been diagnosed, then the odds are higher that other family members are also affected.
Pedigree analysis, which is essentially reviewing a family tree, is very important when a patient has been diagnosed with Fabry disease. In this context, it is important to educate other members of the family—even members of the extended family— as, if one member has been diagnosed with Fabry disease then other family members may too be suffering from the disease.
A diagnosis of Fabry disease is confirmed by a laboratory test. In males, a blood test measures the level of the affected enzyme in the blood. If these levels are reduced in males, Fabry disease can be diagnosed. DNA testing can also be undertaken as further confirmation.
In women, because of the effect of lyonization, the enzyme activity can actually be normal; therefore, genotyping, which is basically the analysis of the gene mutation of alpha-galactosidase A, is vital to confirm the diagnosis in a female patient.

Why can diagnosis of this condition sometimes take a long time?
In Fabry, it can take 20 years, and some literature even quote 40, to obtain a diagnosis of Fabry disease in an individual patient.
The fundamental problem with Fabry disease, —I like to compare it to a chameleon—is that it looks like a lot of other diseases. Because it’s a rare disease, caused by a rare genetic mutation, people don’t consider Fabry disease an option, or they aren’t even aware that Fabry disease exists.
The symptoms of Fabry disease are very unspecific and can be seen in many other conditions. In the classical presentation of Fabry disease, pain is the first symptom, but in the non-classical presentation, which is found particularly in women, the first symptoms could also include a stroke at a young age or very unspecific neurological symptoms that might be misdiagnosed.
Fabry disease patients have been known to have been misdiagnosed with multiple sclerosis, and some children, because of the prolonged episodes of pain and gastrointestinal symptoms, may be diagnosed with rheumatic fever.
The diagnosis is very challenging, and patients often visit a number of doctors and specialists before Fabry disease is finally confirmed.
I think the key to improve diagnosis is if there is a certain symptom or symptom cluster for a prolonged period of time that can’t really be explained properly, to consider that this might be a rare genetic disorder and seek a genetic specialist to make the diagnosis.
How important is awareness of Fabry disease?
Awareness of Fabry disease is vital. Data exists which indicates that potentially up to 85% of patients who suffer from Fabry disease are not actually diagnosed.
Because of the very dramatic morbidity and mortality, cardiovascular risk, heart, kidney and brain damage, and the development of end-stage renal disease, it is very important to educate about this disease.
In addition, as the disease is often diagnosed at end-stage renal failure and patients are already on dialysis, or may even already be transplanted, it is critical to help halt and, ideally, prevent these types of consequences by starting early.
The other part is educating the family members of patients with Fabry disease. As I mentioned, if a patient has been diagnosed, the likelihood that other family members might be affected is very high. It has been estimated that for every patient diagnosed in a family there may be up to five other family members who also have Fabry disease.
Given the nature of the disease, a lot of concern and anxiety exists around the diagnosis, and there are psychological hurdles in discussing this with family members. In some countries, legal hurdles also exist, with confidentiality issues making it challenging to undertake a pedigree analysis.
Awareness about genetic disorders in general, and Fabry disease in particular, is very critical to preventing the long-term consequences of the disease.
What is currently known about the genetic mechanism of Fabry disease?
The lysosomal enzyme alpha-galactosidase A is encoded by a single gene: the GNA gene. This is located on the long arm of the chromosome X in the position Xq22.
One of the challenges patients experience, also related to the clinical symptoms, is that a variety of mutations to this gene exist. To date, more than 400 mutations have been reported. The mutations vary in severity, meaning that they can either result in a total lack of the enzyme, or in a dysfunctional enzyme that doesn’t perform its job properly.
The majority of these mutations are what is known as “private”, which means that the mutation is only found in a single family or just a few families.
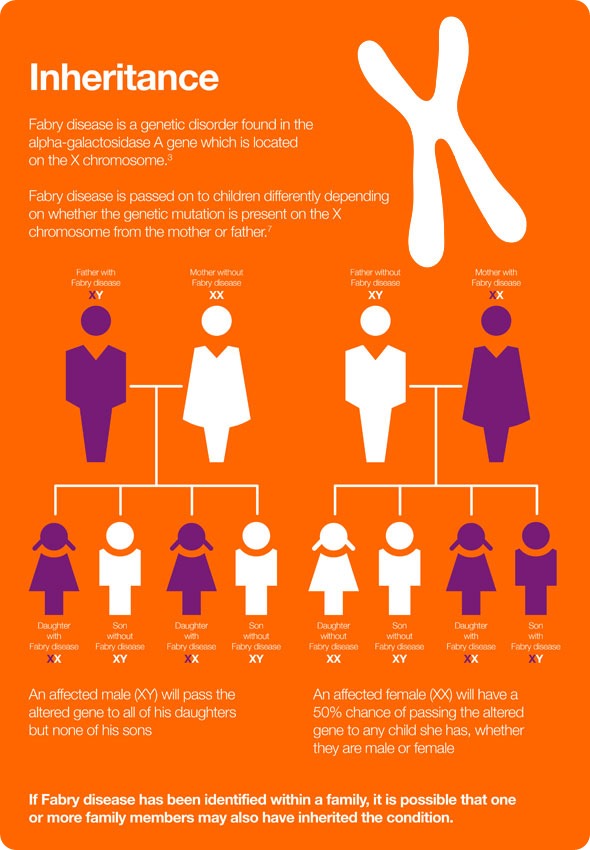
How is Fabry disease inherited?
As the disease is X-linked, an affected male will pass his altered gene to all of his daughters, but none of his sons. An affected female will have a 50% chance of passing the altered gene to any child she has, whether they are male or female.
Historically, females were always said to be carriers of the defective gene and not clinically affected by Fabry disease, but that understanding has now very much been revised and it is now well accepted that women are also impacted by Fabry disease.
It’s a somewhat different clinical picture in that females are clearly affected by Fabry disease, but in a much more variable clinical presentation than males. They’re affected later than males and, fortunately, usually somewhat more mildly.
Females can, of course, also just be carriers and not be currently affected or have any clinical symptoms at all.
What further research is needed to improve our understanding of Fabry disease?
One of the big challenges of Fabry disease is that there’s no good biomarker that allows you to track and predict the severity and the potential progress of the disease, as well as the effectiveness of any therapeutic intervention.
Finding such a biomarker represents a major research need and, given that we are talking about a rare disease, the establishment of a valid biomarker is a challenge in and of itself.
Another challenge, from a research perspective, is to better understand the link between the gene mutation and whether or not this leads to clinical consequences. In other words, there is no good correlation, no good understanding, of how the gene mutation impacts the severity of the clinical symptoms or the onset of the clinical symptoms.
For example, if a diagnosis has been established in an asymptomatic patient, we really don’t know whether the patient is going to develop clinical symptoms at some point in time and whether the patient would benefit from any therapeutic intervention.
The other major area of research we need to address is improvement of the diagnosis: making it easier and cheaper to diagnose. We have seen a lot of development in terms of the technology used for genetic testing, and the costs associated with this have already decreased, but further research is needed to understand how we can diagnose patients earlier and more quickly.
How does Shire plan to help advance innovation in Fabry disease research?
We actually established a program last year that we called Innovating in Fabry. This is a research grant program, where we challenged the scientific community to come up with great ideas that further the scientific understanding of the disease.
The independent research programs, which we will sponsor in this area, revolve around novel ideas that address the significant unmet needs in Fabry disease.
The 2014 grant winners were announced at the beginning of June, and are as follows:
- Derralynn Hughes, MD, United Kingdom - Disease severity and pathophysiology in Fabry disease
- Aleš Linhart, MD, Czech Republic - Cardiac energy metabolism in Fabry disease
- Kathy Nicholls, MD, Australia - Use of stem cells and podocytes to investigate Fabry nephropathy
We look forward to the results of this research because we hope that these projects help further the understanding of Fabry disease.
Where can readers find more information?
About Hartmann Wellhoefer
 Dr. Wellhoefer joined Shire in 2012 and leads the Medical Affairs function for the Rare Diseases portfolio including Fabry, Gaucher, Hunter Disease as well as Hereditary Angioedema.
Dr. Wellhoefer joined Shire in 2012 and leads the Medical Affairs function for the Rare Diseases portfolio including Fabry, Gaucher, Hunter Disease as well as Hereditary Angioedema.
With over 20 years of experience in the pharmaceutical industry, Dr. Wellhoefer has worked in a variety of roles including drug target evaluation, business development, clinical development, life cycle management (LCM) and medical affairs in global, regional and local functions in Europe and the United States.
Dr. Wellhoefer has an MD from Technical University, Munich, Germany, and trained in Internal Medicine at Rechts der Isar, University Hospital also in Munich.
References
- 1. Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd, 2006: Chapter 2.
- Heese BA. Current strategies in the management of lysosomal storage diseases. Semin Pediatr Neurol 2008;15:119-26.
- Rare Disease. About Rare Diseases.
Available at: https://www.raredisease.org.uk/ Last accessed April 2014.
- Mehta A, Widmer U. Natural history of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd, 2006: Chapter 19.
- Marchesoni CL. et al. Misdiagnosis in Fabry disease. J Pediatr. 2010 May;156(5):828-31.
- Desnick RJ. et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 2003;138:338-346.
- Vanier MT and Caillaud C. Disorders of Sphingolipid Metabolism and Neuronal Ceroid-Lipofuscinoses. In: Saudubray J-M, et al. (Editors).
Inborn Metabolic Diseases. 5th edition, 2012. Springer Berlin, Heidelberg.
 New genomic approach links rare immune cells to autoimmune disease risk
New genomic approach links rare immune cells to autoimmune disease risk