Although there have been advances in microsurgical procedures, options for treatment to restore prior function after peripheral nerve injury are still unavailable. Currently, autologous nerve grafting remains the favored therapy.
However, experimental researchers recently have concentrated efforts on artificial constructs development, specifically to combine smart biomaterials and stem cells, with the aim of enhancing the results of nerve autografting.
Chemically stimulated human adipose-derived stem cells (dhASC) are seen to improve nerve regeneration results. However, when chemical stimulation is withdrawn, these properties are lost, and low survival rates upon transplantation have been associated with the procedure. It is theorized that interactions with synthetic hydrogel matrices could preserve and enhance neurotrophic characteristics of dhASC.
dhASC is cultured on PeptiGel-Alpha 1 and PeptiGel-Alpha 2 self-assembling peptide hydrogels, demonstrating similar feasibility to collagen I control gels. Culturing dhASC on Alpha 1 and Alpha 2 substrates permit the neurotrophic features to be maintained. For example, the expression of growth factors and neuroglial markers are both maintained.
Both Alpha 1 and Alpha 2 substrates are appropriate for the culture of peripheral sensory neurons, allowing the sprouting of neuronal extensions, but not requiring biological extracellular matrices while still preserving neuronal function.
PeptiGel substrates with added hdASC are projected to be promising contenders in tissue engineering therapy development for the repair of peripheral nerve injuries.
Introduction
Peripheral nerve injuries (PNI) commonly occur because of upper and lower limb trauma. They greatly affect the quality of life of the patient and can lead to a considerable socioeconomic burden.
Although there has been extensive research in the field of nerve regeneration, as well as noteworthy developments in microsurgical techniques, treatment options currently offered are still limited to end-to-end nerve repair, which utilizes nerve autografting when there is the presence of a nerve gap.
This technique requires a second surgery, leads to donor site morbidity, and continues to lead to disappointing regeneration outcomes and suboptimal motor-sensory functional results. More recently, the development of bioengineered nerve grafts has been of increased interest.
Bioengineered nerve grafts merge the benefits of smart biomaterials and engineered 3D scaffolds with possible gene therapy approaches and pharmacological intervention for a personalized, fully synthetic device for peripheral nerve repair.
Although several commercial products are currently available, there are none in routine clinical use or that have shown superiority to nerve autograft; this may be linked to their inability to account for the biology of the regenerating nerve.
Several substrates have been evaluated as possible nerve guidance tubes in experimental models, including blood vessels, extracellular matrix-based scaffolds, chitosan, alginate or silk fibroin-based tubes, and synthetic polymer-based conduits.
Microtopographical cues, as well as electrospun or 3D printed microchannels, are often featured on synthetic nerve guidance tubes. Failing these options, similar environmental cues can facilitate the alignment of endogenous “repair” cells, in addition to increasing the speed and directing axonal growth toward the distal stump.
A particularly interesting possibility for enhancing the results of nerve repair is the introduction of exogenous cell therapy within the conduit lumen. For this process, it is required that relevant cell types that are easy to harvest and expand are identified and that they present high neurotrophic and regenerative potential.
Schwann cells (SCs) would be perfect candidates due to the role they play during endogenous nerve repair. However, yielding an adequate amount of autologous SCs would lead to donor site morbidity and need extensive culture expansion time.
To establish an alternative source of transplantable cells, many cell types have been evaluated, including adult stem cells from a variety of niches, namely bone marrow, skin, umbilical cord, and dental pulp. These adult stem cells are the most promising alternative.
The current study, along with others, have previously shown that human adipose-derived stem cells can be chemically differentiated in vitro toward a Schwann-like phenotype (hdASC), with enhanced neurotrophic potential, including improved expression of several neurotrophins.
However, it must be noted that this chemically-acquired phenotype speedily reverts after the withdrawal of the differentiation stimulus with loss of neurotrophic features, signifying that an alternative approach for differentiation might be needed. Additionally, reports have shown transplanted cell survival at the peripheral nerve injury site to be low, encouraging an enhanced approach for stem cell delivery.
Hydrogels are reticulated polymer structures categorized as having high water content and versatility of engineering, making them perfect scaffolds for biomedical applications. They are particularly appropriate for peripheral nerve bioengineering.
Hydrogels mimic the environment of the target tissue to favor differentiation, cell survival, and integration to the host tissue. Hydrogels that are derivatives from natural sources are characterized by ideal biocompatibility and biodegradability, but low purity and immune rejection risk are a significant hindrance to clinical translation.
Conversely, excellent control is possible over the purity of synthetic hydrogels, but biocompatibility is frequently a problem. For this reason, there is growing attention paid to the progression of synthetic hydrogels created with naturally occurring molecules, like amino acids, to create a combination of benefits of both natural and synthetic hydrogels.
Short peptide molecules have demonstrated the ability to self-assemble into water-swollen networks creating highly biocompatible and biodegradable hydrogels (SAPs) with optimal properties for biomedical applications.
Self-assembly of SAPs can be accomplished with different designs like coiled-coils, short aromatic peptides, β-hairpin, peptide amphiphiles, and β-sheet systems.
The purpose of this study is to explore the suitability of commercially-available short self-assembling peptide sequences for the culture of hdASC. Hypotheses suggest that the Manchester BIOGEL PeptiGels hydrogels can allow survival and proliferation of hdASC and that the interaction between the cells and the biomaterial will permit the conservation and enhancement of the differentiated neurotrophic phenotype of hdASC.
hdASC would be delivered using PeptiGels within the lumen of a pre-existing nerve guidance conduit, where the enhanced neurotrophic function could be exerted. The progression of a stem cell-delivery platform for autologous hdASC, with the capabilities of enhancing cell survival and differentiation state, could lead to important advances for the development of a fully synthetic bioengineered nerve graft for the treatment of PNI.
Results
Are self-assembling peptide (SAP) hydrogels appropriate scaffolds for the culture of hdASC?
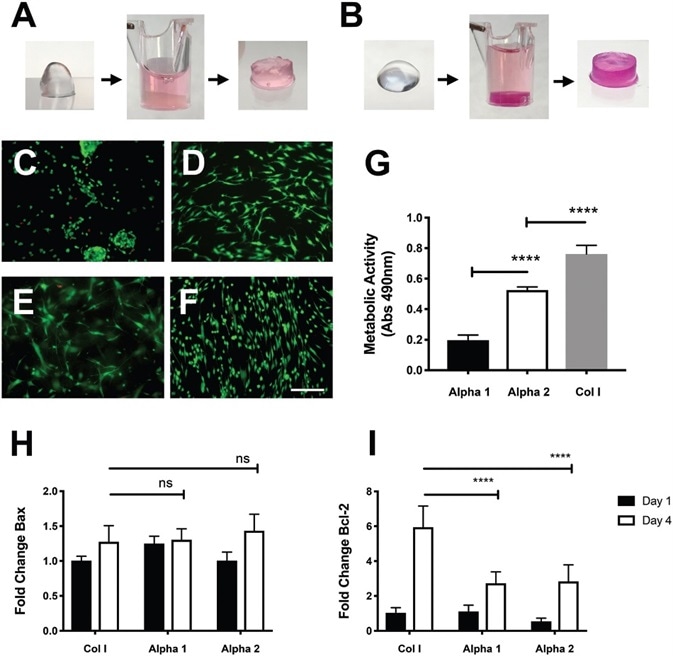
Figure 1. Viability of hdASCs on novel self-assembling peptide hydrogels. A) Alpha 1 and B) Alpha 2 hydrogels prior and after media conditioning. The change of pH causes the hydrogels to set within the cell culture insert, phenol red dye penetrates into the gel causing pink coloration. LIVE/ DEAD staining shows high viability (green cells) of hdASCs on C) Alpha 1 and D) Alpha 2, with negligible numbers of dead cells (red staining). hdASC morphology on Alpha 2 (D) was spindle-shaped, similar to Col I E) and tissue culture plastic controls F); whereas cell morphology on Alpha 1 hydrogels (C) was more rounded, although there was no increase in cytotoxicity, as shown by similar amounts of red-stained dead cells. G) Viability assay performed after 4 d in vitro showed a significantly higher viability of hdASCs on Alpha 2 hydrogels compared to Alpha 1 (****P ∠0.0001, n =3). Viability on control Col I gels was significantly higher when compared to both Alpha 1 and Alpha 2 hydrogels after 4 d of culture (****P ∠0.0001, n =3). H) There was no difference in the expression levels of the proapoptotic gene Bax across all substrates following 4 d of hdASC culture (ns). I) Expression levels of the antiapoptotic gene Bcl-2 were significantly lower on Alpha 1 and Alpha 2 hydrogels after 4 d of culture, compared to Col I substrates (****P ∠0.0001). Scale bars 100 μm. Image Credit: Manchester BIOGEL
Self-assembling peptides, namely, PeptiGel-Alpha 1 and PeptiGel-Alpha 2, were conditioned by incubation in stem cell culture media for a minimum of 2 hours. This permitted gel formation before cell seeding, as shown in Alpha 1, Figure 1A; Alpha 2, Figure 1B. After 4 days of culture, no sign of major cell death was observed on hdASC in either PeptiGels, with levels similar to Col I (control).
Most of the cells were positive for the live staining (Calcein-AM, green, Figure 1 C–F). Remarkably, the cell morphology on Alpha 2, as shown in Figure 1D, was very similar to the spindle-shaped morphology typical of hdASC when cultured on traditional tissue culture plastic substrates (Figure 1F) or Col 1 gels (Figure 1E).
To some extent, this is expected because Alpha 2 shares a similar positive charge to tissue culture plastic, while Col I gels have biologically active cues that have the ability to influence cell morphology.
On the contrary, cells grown on Alpha 1 scaffolds were observed to be more rounded in morphology and often assembled in clusters, even though they did not show signs of excessive cytotoxicity, as shown by the small number of cells positive for the dead staining (Ethidium homodimer-1, red, Figure 1C).
Further confirmation of this was demonstrated by live-cell imaging displaying the attachment of green fluorescent protein (GFP)-labelled hdASC on Alpha 1 and Alpha 2 hydrogels. This was seen during the first 56 hours after seeding, as shown in Video S1, Supporting Information, and in the following 45 hours, as seen in Video S2, Supporting Information.
Additionally, the live cell imaging displayed that the two different hydrogels exert a different response of hdASC, with Alpha 1 permitting superior motility, even after attachment for 96 hours. On the other hand, cells seeded on Alpha 2 hydrogels were seen to be more static and less motile, as shown in Videos S1 and S2, Supporting Information.
The variations in cell morphology and motility patterns propose that Alpha 2 gels permit a better quality of cell adhesion in comparison to Alpha 1 gels. For confirmation, viability tests of hdASC were performed on both Alpha 1 and Alpha 2 scaffolds [3-(4,5-dimethylthiazol-2-yl)- 5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) assay after 4 days in culture].
All hydrogel scaffolds permitted cell attachment and survival, and hdASC demonstrated greater viability on Alpha 2 hydrogels in comparison to Alpha 1 scaffolds.
Alpha 2 mean absorbance at 490 nm ± SD was 0.524 ± 0.021, which was significantly higher than Alpha 1 at 0.1973 ± 0.033 (****P < 0.0001, n = 3, Figure 1G). Viability of hdASC on both Alpha 1 and Alpha 2 hydrogels was significantly lower than the viability reported on Col I gels used as controls (0.763 ± 0.057, ****P < 0.0001, n = 3).
Further investigations were required on the effect of the self-assembling peptide hydrogels on hdASC fate and survival, and so assessment of the expression levels of proapoptotic (Bax) and anti-apoptotic (Bcl-2) genes was completed.
Following 1 day of culture, Bax expression levels were slightly higher on Alpha 1 (*P < 0.05), when compared to Alpha 2 hydrogels (ns), and further compared to Col I control gels, as shown in Figure 1H.
Day 4 displays the expression levels of Bax at significantly increased levels on Col I gels (* P < 0.05) and Alpha 2 hydrogels (****P < 0.0001), but not on Alpha 1 scaffolds (ns) in comparison to the respective day 1 samples.
However, no significant differences in Bax expression levels were shown on day 4 across the different substrates. This is represented in Figure 1H.
Bcl-2 expression levels were comparable across all substrates after 1 day of culture, and no significant differences were seen, as demonstrated in Figure 1I. After four days in culture, Bcl-2 levels on all substrates were significantly increased in comparison to day 1 controls (***P < 0.001 for Alpha 1 and ****P < 0.0001 for Col I and Alpha 2).
Interestingly, Bcl-2 levels at day 4 were significantly lower on both Alpha 1 (****P < 0.0001) and Alpha 2 (****P < 0.0001) hydrogels in comparison to Col I controls. This is displayed in Figure 1I.
Long-term hdASCs cultures adjust the mechanical properties of SAP hydrogels
After evaluating the suitability of SAP hydrogels for hdASC cultures, further investigations were made into the effects of hdASC long-term maintenance on the mechanical properties of the hydrogel scaffolds. This is displayed in Figure 2A.
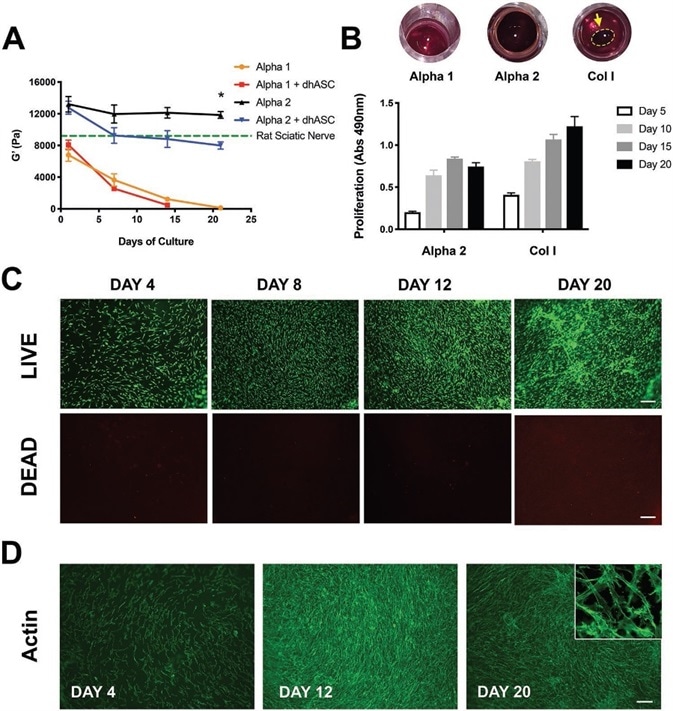
Figure 2. Alpha 2 hydrogels support long-term cultures of hdASCs. A) Rheological studies showed that stiffness of Alpha 1 hydrogels, as measured by storage modulus (G′) rapidly decreases in tissue culture conditions, and when loaded with hdASC the hydrogels are entirely degraded following 2 weeks of culture. The G′ of Alpha 2 was higher and remained stable over a 21 d period (G′ = 11846 ± 433.8 Pa), and was significantly reduced when the gels were loaded with hdASCs (G′ at day 13 = 7988 ± 457.0 Pa, *P < 0.05, n = 3), but remained to stiffness levels similar to those of a rat sciatic nerve (green dotted line). B) Alpha 2 was the only gel allowing steady proliferation of long-term hdASC cultures, whereas Alpha 1 gels were completely dissolved and Col I gels degraded and significantly shrunk, allowing cell growth on the PET membrane underneath the gels and making the assessment of proliferation less reliable. C) LIVE/DEAD staining of hdASC on Alpha 2 showed substantial increase in the number of live cells (green) and negligible amount of cell death (red) over 20 d D) staining for actin filament corroborated the LIVE/DEAD findings and showed the maintenance of the spindle-shape morphology (top right insert in D) typical of differentiated cells. Scale bars 100 μm. Image Credit: Manchester BIOGEL
Rheological analyses demonstrated that in the absence of cells, the storage modulus (G′) of Alpha 2 hydrogels is significantly higher in comparison to Alpha 1 scaffolds (13.2 ± 1.0 kPa vs. 6.7 ± 0.8 kPa, **P < 0.01, n = 3).
In cell culture conditions, there was a progressive decrease in the stiffness of Alpha 1 hydrogels, as revealed by the significant decline of the storage modulus. This came in combination with a decrease in hydrogel thickness with most of the Alpha 1 gels totally degraded after two weeks, and with no gels detectable after 21 days of culture in the presence of hdASC.
Although similar effects were seen in the absence of cells, when the gels were loaded with hdASC, the stiffness reduction and the degradation were accelerated. Remarkably, Alpha 2 hydrogels’ stiffness was well-maintained after 3 weeks of preservation in tissue culture conditions (G′ at day 1 = 13.2 ± 1.0 kPa vs. G′ at day 21 = 11,8 ± 0.5 kPa, ns, P = 0.681) signifying minimal degradation of the gel within this timeframe.
The existence of hdASC sped up the gel degradation with a significantly reduced G′ at day 21 in the Alpha 2 hydrogels loaded with hdASC in comparison to empty hydrogels (G′ = 11.8 ± 0.5 kPa vs 8.0 ± 0.5 kPa, *P < 0.05, n = 3, Figure 2A).
Alpha 2 was the only hydrogel to permit a long-term culture of hdASC, with Col I and Alpha 1 gels degrading and eroding within two weeks, leading to cell growth on the surface of the PET membrane of the tissue culture inserts, rather than on the surface of the hydrogels.
Additionally, Alpha 2 hydrogels stiffness seeded with hdASCs was comparable to the stiffness of a rat sciatic nerve, represented by the green dotted line in Figure 2A.
A metabolic MTS assay, as seen in Figure 2B, was used to measure the proliferation. Results displayed a steady increase in cell proliferation during the long-term cultures up to 15 days on Alpha 2 hydrogels.
This growth curve was comparable to the cell proliferation seen on Col I gels used as controls. However, it is vital to note that by day 10, Col I gels degraded, permitting growth in cells on the PET membrane underneath.
This assumes the assessment of proliferation at days 15 and 20 irrelevant because this is not a real measure of cells grown on the Col I gels.
In panel 2B, the round images highlight the total absence of Alpha 1 hydrogels within the tissue culture inserts at day 20, the strong brown coloration of Alpha 2 hydrogels that continues to cover the whole surface of the inserts, and the decrease of Col I gels to smaller round gels, as represented by the yellow arrow.
The apparent proliferation decrease seen with the MTS assay on Alpha 2 hydrogels at day 20 is most probably a technical fault, rather than an actual drop in cell number. This could be due to the dyed metabolic activity compound being trapped in the gel leading to a reduction in absorbance readout in the supernatant.
For confirmation of this result, LIVE/DEAD staining of hdASC cultures on Alpha 2 was completed at each time point, which displayed a steady increase in the number of live hdASC over time up to 20 days, stained green in Figure 2C, and insignificant amount of dead cells, seen in red.
Further verification of this finding was confirmed through staining of the cytoskeletal protein actin with AlexaFluor488- phalloidin, as seen in Figure 2D, and confocal imaging highlighted the maintenance of a spindle-shaped morphology following 20 days of culture on Alpha 2 (small inset in Figure 2D).
Electron micrographs show hdASC closely interact with SAP hydrogels
For further investigations into the cell–biomaterial interactions, transmission electron microscopy (TEM) of hdASC cultures were performed on Alpha 1 and Alpha 2 hydrogel scaffolds and Col I gels as controls.
On both hydrogels, particularly Alpha 2, hdASC, as depicted by yellow arrows in Figure 3, appeared to interact with gel nanofibers actively, as depicted by red arrows, by sending numerous protrusions throughout the structure of the hydrogel. This is seen in the left column.
At higher magnifications, e.g., 4800× and 9300×, the fine cellular process is increasingly apparent, and caveolae structures could be seen in the plasma membranes (yellow arrows on Alpha2) closely interacting with the peptide nanofibers (red arrows).
Such structures were not identifiable in TEM images of hdASC cultures on collagen gels (Figure 3, bottom row); in Col I gels, fibers were larger in size and more sparse (red arrows).
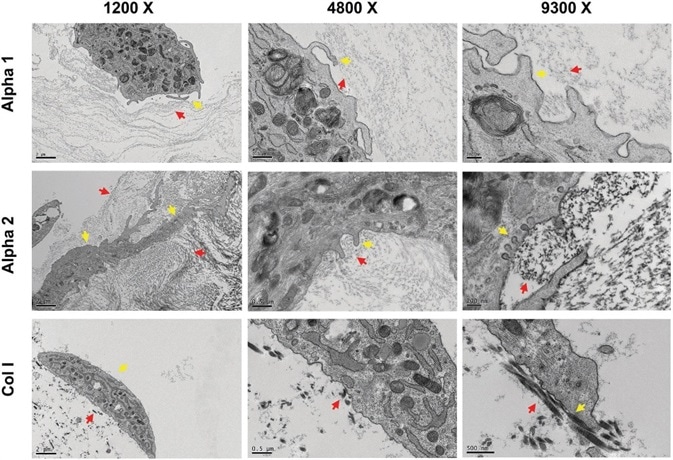
Figure 3. hdASC closely interact with Alpha 1 and Alpha 2 nanofibers. Transmission electron micrograph showed that hdASC (yellow arrows) are actively interacting with the SAPs nanofibers (red arrows) with cells extending processes within the hydrogel structures. At higher magnification (9300×, right panels) it was possible to identify caveolae-like structures, which were not present in control cultures of hdASC on Col I gels (bottom row). Image Credit: Manchester BIOGEL
Culturing on Alpha 1 and Alpha 2 substrates prefers the differentiated state of hdASC
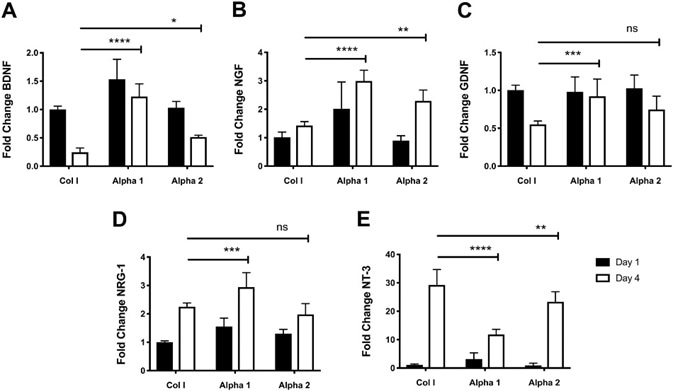
Figure 4. Changes in the expression levels of growth factors indicate an increase of neuroglial differentiation of hdASC on SAPs hydrogels. A) Following 4 d of cultures the expression levels of BDNF were significantly higher on both Alpha 1 (****P < 0.0001) and Alpha 2 (*P < 0.05) hydrogels compared to day 4 Col I controls. B) Similarly, NGF expression levels on Alpha 1 (****P < 0.0001) and Alpha 2 (**P < 0.01) were significantly increased compare to hdASC cultures on Col I at day 4. C) GDNF levels decrease with continued culture of hdASC on Col I gels, but not on Alpha 1 where levels keep significantly higher (***P < 0.001) compared to Col I controls at day 4. The decrease in GDNF expression levels was less striking on Alpha 2, however expression levels at day 4 were not significantly higher than Col I controls (ns). D) Culture of hdASC for 4 d on all substrates caused increase of NRG-1 expression levels; this increase was more noticeable on Alpha 1 hydrogels, which showed significantly higher levels compared to Col I control gels (***P < 0.001). Expression levels of NRG-1 on Alpha 2 after 4 d was comparable to Col I gels. E) NT-3 expression levels were significantly increased after 4 d of cultures on all substrates. The fold increase was significantly lower on Alpha 1 (****P < 0.0001) and Alpha 2 (**P < 0.01) hydrogels compared to Control Col I gels. Image Credit: Manchester BIOGEL
In order to explore the effect of SAPs hydrogels on hdASC phenotype, molecular analyses for common markers of neuroglial differentiation were performed, and comparisons were made on the expression levels to control cultures of hdASC on Collagen hydrogels.
At day 1, expression levels of brain-derived neurotrophic factor (BDNF) were significantly higher on Alpha 1 hydrogels in comparison to Col 1 hydrogels (****P < 0.0001, Figure 4A). However, there were no significant expression changes on Alpha 2 scaffolds (ns, Figure 4A).
After four days of culture on Col I hydrogels, BDNF levels were seen to decrease significantly (****P < 0.0001, Figure 4A). This decrease in BDNF expression was less distinct on both Alpha 1 and Alpha 2 hydrogels.
The fold change of BDNF on Alpha 1 was significantly higher when compared to Col day 1 (****P < 0.0001, Figure 4A). BDNF levels on Alpha 2 hydrogels showed a significant reduction after 4 days of culture, but the expression was still higher in comparison to cultures on Col 1 (*P < 0.05, Figure 4A).
Nerve growth factor (NGF) expression levels were higher on Alpha 1 hydrogels in comparison to control Col I gels after 1 day of culture (***P < 0.001, Figure 4B), whereas expression levels of NGF on Alpha 2 were unaffected (ns).
The 4 days of culture increased NGF expression in all groups in comparison to the respective day 1 controls: Col I (ns); Alpha 1 (***P < 0.001); Alpha 2 (****P < 0.0001).
Interestingly, expression levels of NGF at day 4 on both SAPs hydrogels were significantly higher than Col I day 4 cultures (****P < 0.0001 and **P < 0.01 for Alpha 1 and Alpha 2, respectively, Figure 4B).
Glial cell-derived neurotrophic factor (GDNF) expression levels on all three substrates were comparable after 1 day of culture, as shown in Figure 4C. A significant decrease in GDNF expression was seen in hdASC cultured for 4 days on Col 1 gels (****P < 0.0001) and on Alpha 2 (**P < 0.01), but not Alpha 1 (ns).
The noteworthy point is that GDNF expression levels after 4 days of cultures were significantly higher when cells were cultured on Alpha 1 hydrogels in comparison to Col I controls (***P < 0.001, Figure 4C).
Following 1 day of culture, neuregulin 1 (NRG-1) levels on Alpha 1 hydrogels were significantly higher in comparison to Col I controls (**P < 0.001, Figure 4D). Likewise, NRG-1 expression on Alpha 2 hydrogels was increased, but this was not statistically significant.
Extended culture for 4 days on all substrates showed a significant increase of NRG-1 expression levels in comparison to the respective day 1 controls: Col I (****P < 0.0001), Alpha 1 (****P < 0.0001), and Alpha 2 (***P < 0.001).
A more distinct increase in NRG-1 expression was observed when cells were cultured on Alpha 1 with levels significantly higher than Col I day 4 cultures (***P < 0.001, Figure 4D). Remarkably, NRG-1 expression levels of hdASC cultured for 4 days on Alpha 2 hydrogels were comparable to Col I cultures.
Expression levels of neurotrophin 3 (NT-3) following 1 day of cultures were comparable for all three substrates with no observations of statistical significance or differences between groups, as shown in Figure 4E.
After 4 days of culture, increases in NT-3 levels were witnessed for all substrates in comparison to day 1 controls: Col I (****P < 0.0001, n = 3); Alpha 1 (****P < 0.0001), Alpha 2 (****P < 0.0001). It is interesting to note that NT-3 levels were significantly lower on both Alpha 1 (****P < 0.0001) and Alpha 2 (**P < 0.01) in comparison to Col I at day 4 (Figure 4E).
Short-term hdASC culture over one day on Alpha 1 created a significant increase in the expression of tropomyosin receptor kinase A (TrkA) in comparison to Col I control gels (****P < 0.0001, n = 3, Figure 5A).
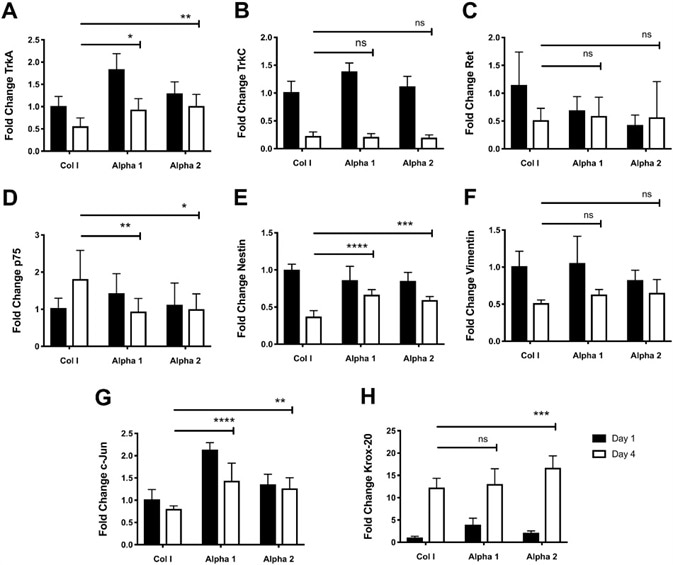
Figure 5. Culture of hdASC on SAPs hydrogels alters expression of growth factor receptors, transcription factors, and cytoskeletal molecules. A) Following 4 d of cultures, TrkA expression levels were significantly higher on both Alpha 1 (*P < 0.05) and Alpha 2 (**P < 0.01) hydrogels compared to day 4 Col I controls. B) TrkC expression levels were higher on Alpha 1 hydrogels at day 1 compared to Col I controls; however by day 4 there were no differences in TrkC expression levels across all groups. C) Similarly, expression levels of GDNF receptor Ret were lower on both Alpha 1 and Alpha 2 hydrogels compared to Col I controls at day 1; nevertheless, by day 4 there were no differences in expression levels of Ret across all groups. D) p75 expression levels were significantly lower on Alpha 1 (**P < 0.01) and Alpha 2 (*P < 0.05) hydrogels compared to Col I gels at day 4. E) Expression of Nestin was significantly higher on Alpha 1 (****P < 0.0001) and Alpha 2 (***P < 0.001) hydrogels compared to Col I controls at day 4. F) Expression levels of the intermediate filament Vimentin after 4 d of hdASC culture on all substrates was comparable and no significant differences were observed. G) c-Jun expression levels at day 4 were significantly higher on Alpha 1 (***P < 0.001) and Alpha 2 (**P < 0.01) hydrogels compared to day 4 Col I scaffolds. H) Krox-20 expression levels increased with prolonged hdASC culture in all substrates; interestingly, at day 4 Krox-20 expression was significantly increased on Alpha 2 (***P < 0.001) compared to Col I controls. Image Credit: Manchester BIOGEL
The same effect was not witnessed on Alpha 2 gels (ns vs. Col I day 1). Following 4 days in culture, a significant decrease of TrkA expression was observed on Col I gels (**P < 0.01), on Alpha 1 hydrogels (****P < 0.0001), however, not on Alpha 2 substrates (ns).
Of note, although significant decreases in TrkA expression levels were observed on Alpha 1 substrates, these continued to levels similar to Col I gels at day 1. Indeed, TrkA expression after 4 days was significantly higher on both Alpha 1 (*P < 0.05) and Alpha 2 (**P < 0.01) hydrogels in comparison to Col I controls at day 4.
Tropomyosin receptor kinase C (TrkC) expression levels on Alpha 1 after one day of cultures showed significant increases in comparison to Col I gels (****P < 0.0001, Figure 5B).
However, after 4 days of culture, the expression of TrkC was significantly decreased across all groups (****P < 0.0001, Figure 5B). Expression of the GDNF receptor Ret following 1 day of culture was lower on both Alpha 1 (ns) and Alpha 2 (**P < 0.01) hydrogels in comparison to Col I controls, as shown in Figure 5C.
Extended culture of hdASC on Col I gels for 4 days lead to a significant decrease in Ret expression levels (*P < 0.05). Decreases were not seen in ret expression levels on either Alpha 1 (ns) and Alpha 2 (ns) hydrogels in comparison to the respective day 1 samples and were similar to Col I day 4 controls, as shown in Figure 5C.
Comparable expression levels of p75 were observed after one day of culture across all groups, as shown in Figure 1D. Following 4 days, p75 expression levels were significantly increased on Col I gels (*P < 0.05), but the same was not seen on Alpha 1 and Alpha 2 hydrogels.
p75 levels were significantly lower at day 4 on Alpha 1 (**P < 0.01) and Alpha 2 hydrogels (*P < 0.05) in comparison to day 4 Col I controls, as seen in Figure 1D.
Nestin, an intermediate filament protein vital for the development of the nervous system development showed expression levels slightly lower on Alpha 1 (ns) and Alpha 2 (*P < 0.05) scaffolds in comparison to Col I gels after 1 day of culture, as shown in Figure 5E.
After 4 days of cultures, expression levels of Nestin were significantly decreased on all substrates (****P < 0.0001, **P < 0.01 and ****P < 0.0001 for Col I, Alpha 1 and Alpha 2, respectively). However, expression on Alpha 1 (****P < 0.0001) and Alpha 2 (***P < 0.001) hydrogels continued to be significantly higher when in comparison to day 4 Col I cultures (Figure 5E).
On day 1, Vimentin, an intermediate filament protein with main roles for cytoskeleton integrity showed expression levels comparable on Col I gels and Alpha 1 hydrogels (ns), and slightly decreased on Alpha 2 scaffolds (ns, Figure 5F).
Significant decreases of Vimentin expression levels were seen overtime on Col I gels (****P < 0.0001), Alpha 1 hydrogels (***P < 0.001), but not on Alpha 2 scaffolds (ns). The reason behind this was because, at day 1, Vimentin was already lowly expressed on Alpha 2 hydrogels.
For all substrates, the expression levels at day 4 were similar, as shown in Figure 5F. The expression levels of c-Jun after 1 day of culture were significantly higher on both Alpha 1 (****P < 0.0001) and Alpha 2 (*P < 0.05) hydrogels, in comparison to control Col I gels (Figure 5G).
Following four days, although there were very slight decreases, the levels of c-Jun continued to be significantly higher on Alpha 1 (****P < 0.0001) and Alpha 2 (**P < 0.01) hydrogels, in comparison to Col I control gels (Figure 5G).
On day 1, the transcription factor Krox-20 showed an expression that was significantly higher on Alpha 1 (*P < 0.05), but not on Alpha 2 (ns) hydrogels, in comparison to Col I controls, as shown in Figure 5H.
When hdASC was cultured on all substrates for 4 days, Krox-20 expression levels significantly increased (all ****P < 0.0001 in comparison to respective day 1 controls). Interestingly, Krox-20 expression levels on Alpha 2 hydrogels was significantly higher than the expression seen on control Col I gels (***P < 0.001).
No significant differences were seen in the expression levels of Krox-20 on Alpha 1 hydrogels (ns) in comparison to Col I gels at day 4 (Figure 5H).
Attachment and sprouting of neurites on dorsal root ganglia neurons on Alpha 1 and Alpha 2 hydrogels without the need for biological ECM coating
After evaluating the suitability of SAPs hydrogels for hdASC culture with specific attention paid to stem cell delivery in nerve injury settings, investigations continued regarding their effect on neuronal attachment, survival, and neurite outgrowth, which are all vital requirements for effective nerve regeneration.
Dissociated rat dorsal root ganglia (DRG) neuron cultures were received and maintained for 72 hours on Alpha 1 and Alpha 2 scaffolds and Col I gels (controls) with and without a laminin precoating.
Interestingly, both Alpha 1, as seen in Figure 6A, and Alpha 2, as seen in Figure 6B, substrates permitted attachment of DRG neurons and neurite sprouting with no need for laminin coating.
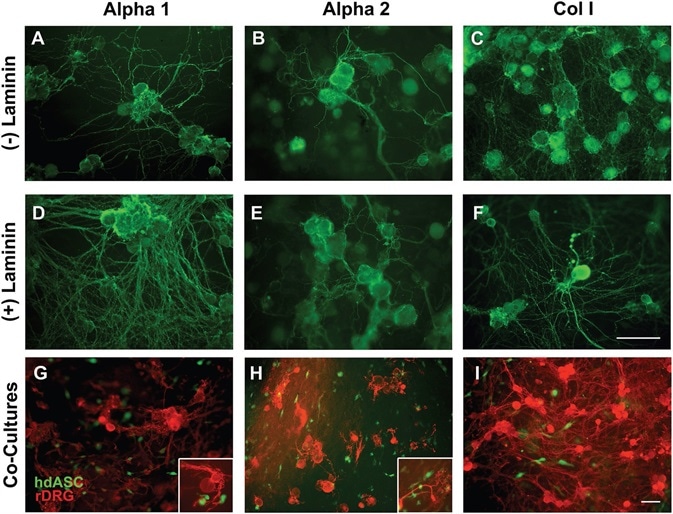
Figure 6. Alpha 1 and Alpha 2 are suitable substrates for neuronal cultures allowing neurite outgrowth without the need of biological ECM coatings. A–C) Rat DRG neurons successfully attached to Alpha 1 and Alpha 2 hydrogels, without any need of laminin coating, which is normally a requirement for any nonbiological substrate. A dense network of neurites was observed on both SAPs and on Col I control gels. D–F) When laminin precoating was applied, the density of the neurite network was increased on all substrates. G–I) Successful cocultures of hdASC and rat DRG neurons were established on all substrates, and close interactions between neuronal extensions and hdASC were observed (see higher magnification inserts). Scale bars 100 μm. Image Credit: Manchester BIOGEL
Alpha 1 permitted greater neurite length and branching in comparison to Alpha 2. When an application of a coating of extracellular matrix was used before cell seeding (Figure 6D–F), the neurite network was denser in comparison to bare substrates.
Finally, coculture experiments were performed in which rat DRG neurons were plated on substrates that were pre-seeded with GFP-labeled human hdASCs.
DRG neurons effectively attached to Alpha 1 (Figure 6G) and Alpha 2 (Figure 6H) hydrogels and sprouted neurites that made positive contacts with the pre-seeded hdASCs (see insets in Figure 6 panels G and H for higher magnification images); this was comparable to that seen on the biological extracellular matrix molecules (ECM) Col I, as shown in Figure 6I.
Functionally active DRG eurons on Alpha 1 and Alpha 2 substrates
For confirmation to verify that DRG neurons hold functionality when cultured on Peptigels, cytosolic Ca2+ concentration ([Ca2+]i ) was measured after stimulation with 50 × 10−3 M KCl. DRG neurons were seeded on Alpha 1 and Alpha 2 hydrogels and permitted to sprout neurites over 48–72 hours, as shown in Figure 7A.
![DRG neurons elicit action potentials when cultured on Alpha 1 and Alpha 2 hydrogels. A) DRG cultures were established on Alpha 1 (left) and Alpha 2 (right) and allowed to sprout neurites for 72 h. B) Cells were loaded with Fluo-4 and following KCl 50 × 10− 3 challenge C) they showed an increase in Ca2+-dependent intracellular fluorescence (yellow arrows). D) Representative high magnification images of DRG neurons before (left) and during (right) KCl stimulation and corresponding trace showing an increase in [Ca2+]i measured within the regions of interest (red dotted circles). Scale bars 100 μm.](https://www.news-medical.net/image-handler/picture/2020/2/Art1-7.jpg)
Figure 7. DRG neurons elicit action potentials when cultured on Alpha 1 and Alpha 2 hydrogels. A) DRG cultures were established on Alpha 1 (left) and Alpha 2 (right) and allowed to sprout neurites for 72 h. B) Cells were loaded with Fluo-4 and following KCl 50 × 10−3 challenge C) they showed an increase in Ca2+-dependent intracellular fluorescence (yellow arrows). D) Representative high magnification images of DRG neurons before (left) and during (right) KCl stimulation and corresponding trace showing an increase in [Ca2+]i measured within the regions of interest (red dotted circles). Scale bars 100 μm. Image Credit: Manchester BIOGEL
Following loading the cultures with Fluo4-AM (Figure 7B), cells were tested with KCl 50 × 10−3 M to lead to membrane depolarization, induce action potentials, and to create inward calcium influx.
Several DRG neurons on both hydrogel substrates replied to KCl challenge with a prominent increase in [Ca2+]i, as depicted by yellow arrows in Figure 7C. The increase in [Ca2+]i was fast and followed by recovery, as shown in the Videos S3 and S4 (Supporting Information).
The time course of [Ca2+]i dynamics, evaluated from higher magnifications images, is shown in Figure 7D (regions of interests, red dotted circles, were positioned within the DRG cytoplasm).
Discussion
Functional outcomes for PNI patients validate the requirement for a change in clinical management. Therapies that are currently available are limited to surgical repair with unsatisfactory outcomes, have a major impact on the quality of life of patients, and have high costs socioeconomically.
As with several other fields, stem cell therapies signify a new and promising method for the treatment of PNI.
Particularly, adipose-derived stem cells are plentiful, easily expandable, and exhibited great promise in nerve regeneration improvements in a variety of in vitro and in vivo models. However, many disadvantages are significantly delaying the path able to take ASC therapies into clinical use.
Despite the increasing body of experimental literature supporting the effectiveness of ASC intervention, serious uncertainties persist concerning ASC survival after transplantation in vivo, and of ASC regenerative phenotype maintenance.
Reports show survival rates of transplanted cells at the injury site of a damaged nerve to be as little as 0.5%, and two weeks following transplantation ASCs are undetectable in the regenerating nerve.
It is suggested that this is due to the hostile environment of the injury milieu with inflammatory molecules in abundance and the occurrence of high concentrations of adenosine triphosphate, which has been revealed to induce cell death via P27X receptors in ASCs and Schwann cells.
As a method to enhance the survival of cells, continued research is focusing on designing optimized cell delivery approaches, including novel smart biomaterials.
Stem cells are required to be delivered to the site of injury for peripheral nerve repair and effort is being made to imitate the environment of an injured nerve in terms of chemical properties (ECM environment, bioactive moieties, and charge), and mechanical characteristics (stiffness, physical structure, and degradation profile).
This study examined the suitability of SAP hydrogel substrates to cultivate ASC and neuronal cells in order to exploit these novel biomaterials for the engineering of nerve tissues.
Previously, SAP hydrogels have been used for similar applications. RADA16-I, also known as BD Puramatrix, is a β-sheet SAP that has been effectively used to deliver SCs within membrane conduits to repair 10 mm rat sciatic nerve gaps, resulting in improved regeneration distances.
More recently, RADA-16 has been successfully functionalized for improved regeneration outcomes with functional moieties such as IKAV, RGD, BMHP1, demonstrating the versatility of this approach.
Additionally, RADA-16 has been used successfully to encapsulate hASC myocardial infarction models in vivo. RADA-16 hydrogels have many drawbacks linked to the 16 amino acid chain, including the occurrence of arginine in the peptide backbone and the acidity (pH 3.5), which is harmful to cell survival and the complexity of synthesis, increasing production costs.
This article examined a family of shorter amphipathic β-sheet forming peptides, categorized by the alternation of hydrophilic and hydrophobic residues within the peptide backbone, which imitates the components of biological ECM like collagen.
Specifically, we tested a soft (G′ ≈ 8 kPa) hydrogel with a neutral charge (Alpha 1) and a stiffer (G′ ≈ 14 kPa) gel with a positive charge (Alpha 2). hdASC was successfully cultured on both hydrogel types exhibiting a good survival rate that was compatible with Col I control gels and an insignificant amount of cell death.
Although the cells’ metabolic activity (evaluated using MTS analysis) was lower on Peptigels after 4 days of culture in comparison to Col I gels, an increase in cell death was not detected, and the expression of proapoptotic genes (Bax) was comparable to controls.
The gene expression levels of antiapoptotic gene Bcl-2 was lower after 4 days of culture on Peptigels in comparison to Col I gels, which could help elucidate the lower metabolic activity.
This did not, however, result in increased apoptosis cell death as measured by Live-Dead assay, and did not affect the ability of hdASC to proliferate on Peptigels. There is an increased likelihood that the lower levels of antiapoptotic gene Bcl-2 on Peptigels are adequate to counteract the proapoptotic genes effects (e.g., Bax).
It is also vital to note that the expression of these genes was evaluated only at mRNA and not at the protein level and that there are other pro- and anti-apoptotic genes that are yet to be investigated.
Remarkably, striking differences were observed in cell response between the two Peptigel hydrogels. The live imaging studies showed greater motility in cells with a rounded morphology on Alpha 1, while on Alpha 2, the typical spindle-shaped morphology was retained, which is usually seen on tissue culture plastic, and proven a decreased level of motility in the gel.
These specific responses could be due to the stiffness differences between the two substrates. The stiffer Alpha 2 hydrogel resembled the behavior of hdASC on cell culture plastic, while the softer Alpha 1 cells’ hydrogel assumed a rounded morphology more characteristic of nonadherent cells.
However, it is speculated that these effects are more likely to be due to the differential charges between the two substrates. When compared to treated tissue culture plastic, Alpha 2 is positively charged, which is a property known for augmenting cell adhesion and proliferation.
This article supports the likelihood that this is more likely to be the critical difference between the two gels, because there is only marginal variance in stiffness, and the effect of the charge is often described to override mechanical properties. A synergistic effect between charge and stiffness, leading to the changes seen in cell behavior, cannot be excluded.
Hydrogel and matrix stiffness can have effects on cell proliferation, motility, and differentiation phenotype. Rheological studies here suggested that after 20 days of hdASC culture, the stiffness of Alpha 2+cells composite is comparable to rat peripheral nerve.
On the contrary, Alpha 1 gels were completely degraded after 10 days of culture, and Col I control gels had shrunk significantly, permitting the cells to migrate out and proliferate on the membrane of the cell insert.
These results suggest that only Alpha 2 is appropriate for long term cultures of hdASC, and Alpha 2 could be the ideal substrate for delivery of dhASC when precise cell localization is needed after cell transplantation, as seen in PNI.
Additional tests are needed to evaluate this in vivo, but the potential to retain the hdASC at the injury site in the first 2–3 weeks after repair is one of the prerequisites for successful nerve regeneration in nerve repair approaches with the involvement of bioengineered nerve grafts.
hdASC has a neurotrophic profile that permits them to deliver growth factors at the injury site that have been shown to enhance regeneration results. However, there is instability in chemical differentiation, and it cannot be sustained if any of the stimulating agents are taken away.
Due to this reason, it is vital that after delivery of the cells, the induced neuroglial neurotrophic phenotype is maintained.
Yet, the concomitant delivery of the differentiation cocktail and transplanted cell bring significant clinical problems. This study showed close interactions between the gel nanofibers and hdASC by electron microscopy.
This is suggestive of close cell-biomaterial interactions leading to changes in cell phenotype. Gene expression analyses were performed for many markers of hdASC differentiation like BDNF, NGF, GDNF, and Nestin.
After 4 days of cultures on Peptigels, hdASC showed similar or significantly higher levels of these main differentiation markers signifying that the neuroglial phenotype is retained and the neurotrophic potential is not lost.
Similarly, genes like p75 and NT-3, which are synonymous with dedifferentiated and immature SC phenotype, were significantly lower after 4 days of culture on Peptigels in comparison to Col I control gels. This suggests an enhanced differentiated state of dhASC on PeptiGel.
Gene expression levels of c-Jun were also analyzed, and are the main transcription factor in SC biology, which is linked with SC repair phenotype. c-Jun gene expression levels were significantly higher on both Alpha 1 and Alpha 2 gels after 4 days of cultures when compared with Col I control gels.
Higher levels of c-Jun are important for the induction of the repair program and for the preparation of the SC to remyelinate the regenerating axons.
After the nerve repair is complete, it is expected that the SC redifferentiate and form myelin, which is driven by a distinct set of growth factors inclusive of Krox-20. This study discovered that the culture of hdASC for 4 days on Alpha 2 gels significantly increases expression levels of Krox-20 in comparison to Alpha 1 and Col I control gels.
This is particularly relevant because Alpha 2 is the only gel permitting long term culture of hdASC up to 20 days, which is when the remyelinating phenotype is most probably going to be required.
As far as it is known, this is the first study in which the phenotype of ASC was examined in detail after culture on SAPs hydrogels. Previous studies showed limitations in the analysis of a limited set of genes involved in angiogenesis, for example, vascular endothelial growth factor and hepatocyte growth factor.
A significant note is made relating to this article, which analyzed the expression of key markers of differentiation only at the mRNA level, but protein expression levels were not assessed, constituting a limitation of this research.
After assessing how compatible Peptigels are for hdASC long term culture, expansion, and maintenance of differentiated phenotype, this study focused on the suitability of the SAPs for the culture of neurons, which need to regenerate through the PeptiGels-hdASC composites.
Three- to five-day cultures of rat DRG neurons were performed, which commonly require specific ECM requirements for adhesion and neuronal sprouting.
Remarkably, even in the absence of biological ECM (laminin or collagen), DRG neurons had the ability to attach and spread neurite extensions on both Alpha 1 and Alpha 2 gels.
Additionally, when co-cultured with human dhASC, close neuron-glia interactions could be evaluated. Neuronal cultures were viable and were able to elicit action potential as measured by calcium imaging experiments.
The combination of this research confirms the suitability of Peptigels for the culture of peripheral nervous system neurons and glial cells for the first time, suggestive of Peptigels being promising candidates as delivery vectors for hdASC in nerve repair approaches.
The success of the encapsulation of hASC in SAPs gels has been proven for many applications like myocardial infarction and bone regeneration. In relation to peripheral nerve repair, ASCs have been effectively encapsulated in fibrin and cryopolymerized gelatin gels, but there is limited evidence of encapsulation within SAPs.
There are many benefits of SAPs hydrogels for regenerative medicine applications, like low immunogenic profile and potential fine-tuning of the physical, chemical, and mechanical properties. For instance, the addition of graphene oxide flakes can enhance the mechanical strength and conductivity of SAPs, as seen in polyethylene glycol-based gels.
Approaches using a combination of the effect of cell–biomaterial interactions with electrical stimulation could be exploited. An additional benefit of SAPs hydrogels is that protocols are accessible for the extraction of the encapsulated cells for analyses further downstream analyses.
Overall, results of this study suggest that Peptigels substrates, particularly Alpha 2, have excellent potential as delivery vehicles for hdASC in the peripheral nerve injury setting by permitting cell adhesion, cell growth, and maintenance of differentiated properties, as well as allowing neuronal sprouting and function.
These characteristics make Peptigels capable candidates as cell-delivering fillers for pre-existing nerve conduits, including topographical cues. However, further studies are required to be able to assess the feasibility of Peptigels for nerve repair in vivo.
The results here show, for the first time, the effect of cell-biomaterials interaction on the differentiated state of hdASC with enhanced properties for peripheral nerve regeneration.
Experimental section
Preparation and Conditioning of Hydrogel: Synthetic self-assembling peptide hydrogels were attained from Manchester Biogel (Alderley Edge, UK), and additional information on material characterization can be attained from the company.
Two gel types were evaluated, characterized by different charge, and stiffness: PeptiGel-Alpha 1 (G′ ≈ 8.0 kPa, neutral charge) and PeptiGels-Alpha 2 (G′ ≈ 14 kPa, positive charge).
Both gels have a similar stiffness to peripheral nerve tissue and were chosen specifically to examine the effects of stiffness and charge on cell adhesion, proliferation, viability, and differentiation.
Gels were pulse-centrifuged to take out any air bubbles, and 100 µL of each gel was dispersed via a positive displacement pipette (Gilson Scientific, Dunstable, UK) in Thincert cell culture inserts with 1 µm polyethylene terephthalate membranes (Greiner Bio-One, GmbH, Kremsmünster, Austria).
The inserts consisting of the hydrogels were put in 24 well plates and spun at 3000 rpm two times in a plate centrifuge, provided by IEC CL31R Multispeed, Thermo Scientific, UK, for 5 seconds (rotation of the plates occurred at 180 degrees between every spin, allowing flattening of the hydrogel surface).
Peptide hydrogels were conditioned by adding α-Minimum Essential Medium Eagle (αMEM, Sigma-Aldrich, Poole, UK) containing 10% (v/v) Foetal Bovine Serum (FBS, LabTech, Uckfield, UK), to the wells at 2 × 10−3 M L-Glutamine and 1% (v/v) penicillin-streptomycin solution (both Sigma-Aldrich, Poole, UK).
Incubation of plates occurred for a minimum of 1 hour at 37 °C, 5% CO2; media was aspirated and changed with fresh αMEM every 20 minutes before cell seeding. Prepared hydrogels were used on the day or within 24 hours.
Collagen I bovine gels (controls) were organized according to manufacturer’s direction (Col I, Thermo Scientific, UK) and the 5 mg mL Col I stock solution was briefly diluted at a final concentration of 3 mg mL in ice-cold sterile phosphate-buffered saline solution (PBS, Sigma-Aldrich, Poole, UK) enhanced with 15 × 10−3 M NaOH to reach ideal pH (6.5–7.5).
Collagen gels were cast alongside Peptigels and washed with cell culture media before cell seeding. Depending on the assay to be completed, a suitable number of cells (see each Experimental Section for details) was resuspended in 400 µL of media and seeded on the top of the gel to create 2D cultures. Then, an additional 800 µL of media was added to the well at the bottom of the inserts.
Harvest of Adipose-Derived Stem Cell and Differentiation to Neuroglial Lineage: Human adipose-derived stem cells (hASC) were attained from compliant female patients undertaking reconstructive surgery at the University Hospital of South Manchester. All methods had the approval of the National Research Ethics Committee (NRES 13/SC/0499).
Abdominal fat pads were treated as described previously. The freshly isolated adipose tissue was briefly mechanically and enzymatically dissociated with 0.2% (w/v) collagenase I (Life Technologies Ltd, Paisley, UK), dissolved in Hank’s Balanced Salt Solution (HBSS, Sigma-Aldrich, Poole, UK), for 60 minutes at 37 °C.
After filtration with a vacuum-assisted 100 µm mesh (Steriflip, Merk Millipore Ltd, Watford, UK), the stromal vascular fraction was pelleted by 10 minutes of centrifugation at 300 g and resuspended for 1 minute in Red Blood Cell Lysis Buffer (Sigma-Aldrich, Poole, UK).
Following this, a further centrifugation step of 300 g for 10 minutes was completed before resuspension of the cell pellet in αMEM (SigmaAldrich, Poole, UK), 10% (v/v) FBS (LabTech, Uckfield, UK), 2 × 10−3 M L-Glutamine and 1% (v/v) penicillin-streptomycin solution (both SigmaAldrich, Poole, UK).
Cells were preserved in T75 flasks from Corning Ltd, UK, at 37 °C, 5% CO2, with changes in media occurring three times a week and split when subconfluent. Cells were characterized for common cell surface markers, and multipotency was evaluated with general differentiation assays toward adipo-, osteo-, and condro-genic lineages.
At passage 1–2, hASC plated at 30% confluency were chemically induced to a Schwann-like phenotype (hdASC) utilizing a recognized procedure containing three steps: 1) 24-hour treatment with 1 × 10−3 M β-mercaptoethanol (Sigma-Aldrich, Poole, UK); 2) rinsed with HBSS after 72 hours treatment with 35 ng mL all-trans-retinoic acid (Sigma-Aldrich, Poole, UK); 3) 14 days of exposure to a differentiation cocktail containing αMEM supplemented with 5 ng mL platelet-derived growth factor (Peprotech EC Ltd., London, UK), 10 ng mL basic fibroblast growth factor (Peprotech EC Ltd., London, UK), 14 × 10−3 M forskolin (SigmaAldrich, Poole, UK) and 192 ng mL glial growth factor-2 (GGF-2, Acorda Therapeutics Inc., Ardsley, NY, US).
Days 4 and 10 consisted of exposure to the differentiation cocktail, and cells were split and replated in 75 cm2 flasks (2 × 105 cells per flask) for permission of expansion and differentiation. hdASC phenotype at day 14 and onward was confirmed as explained previously and differentiated cells were used for up to 10 passages.
Hydrogels Biocompatibility and Cell Viability Assessment: To valuate biocompatibility of the self-assembling peptides for cell attachment, survival, and proliferation, a sequence of metabolic and immunocytochemical assays were completed.
Viability Assay: hdASC were seeded on Alpha 1, Alpha 2, and Col I gels at a concentration of 1 × 105 cells per well (n = 3). After 3 days of culture, the supernatant was aspirated with care, and the gels were washed with phenol-free Dulbecco’s Modified Eagle’s Medium (DMEM) before incubation in a 20% (v/v) solution of CellTiter 96 AQueous One Solution (Promega, Southampton, UK) diluted in phenol-free DMEM.
This assay is created due to an MTS, which is reduced by cells into a formazan compound, soluble in tissue culture media, and absorbing at 490 nm comparably to the original cell number.
After 90 minutes of incubation at 37 °C and 5% CO2 in the dark, supernatants were moved into 96 well plates, and absorbance at 490 nm was logged utilizing an Asys UVM340 microplate reader/spectrophotometer (Biochrom, Cambridge, UK). Results were expressed as absorbance at 490 nm ± standard error of the mean (SEM, n = 3).
LIVE/DEAD Viability/Cytotoxicity Staining: Cell viability and cytotoxicity were evaluated utilizing LIVE/DEAD Viability/Cytotoxicity Kit according to the manufacturer's direction (Thermo Scientific, UK). An overall of 2 × 104 hASCs was seeded on hydrogel matrices and cultured in standard conditions.
After 4, 8, 12, and 20 days of expansion in vitro, media was aspirated with care, cells were washed with warm PBS and staining solution (2 × 10−6 M calcein-AM and 4 × 10−6 M Ethidium homodimer-1 diluted in PBS) was added on the top of the gels before 15 minutes of incubation at 37 °C.
After the incubation period, gels were taken out of the insert, placed on microscope glass slides, and an Olympus IX-51 fluorescent microscope (Olympus, UK) was used for imaging. For confocal analysis, a Nikon C1 confocal microscope system (Nikon, Japan) was used to image gels.
Cell Proliferation Assay: To evaluate the long-term proliferation potential of hdASC on hydrogel substrates, 2 × 104 cells were seeded, and viability assay was completed as described previously (CellTiter 96 AQueous One Solution kit, Promega) after 5, 10, 15, and 20 days of expansion in vitro. Data were expressed as absorbance at 490 nm ± SEM, n = 4.
Phalloidin staining: Using an identical experimental setup as previously mentioned, assessment of growth in cell number and changes in morphological features were also done by staining for cytoskeletal proteins. After seeding at days 4, 12, and 20, cells were fixed for 30 minutes in 4% (w/v) paraformaldehyde (PFA) in PBS at room temperature.
After rinsing twice with PBS, cells were permeabilized for 30 minutes at room temperature in 0.2 (v/v) % Triton-X 100 and blocked in 1% (w/v) bovine serum albumin (both from SigmaAldrich, UK) and prepared in PBS.
Staining was completed by incubating for 20 minutes at room temperature with AlexaFluor488 phalloidin (1:40, Thermo Scientific, UK). Stained gels were rinsed with PBS two times, taken out of inserts, positioned on glass microscope slides, and imaged with an Olympus IX-51 fluorescent microscope (Olympus, UK).
Rheology: To determine the mechanical characteristics of peripheral nerve tissue, sciatic nerve samples were collected from Sprague Dawley rats at three months old. Terminated of rats occurred according to schedule 1 procedures of the UK Animals (Scientific Procedures) Act 1986.
Skin and muscle in the hind limbs were removed using fine forceps to expose the sciatic nerve, which was separated from surrounding tissues, dissected, and placed in HBSS solution to be stored before rheological measurements.
For determination of mechanical properties of the gel before long-term cultures 6 × 104 hdASC were plated in triplicate on the hydrogel substrates, and rheological measurements were recorded at days 1, 7, 14, and 21.
Measurements were completed in parallel on gels stored in tissue culture conditions with no addition of hdASC as controls. All rheological studies were done using a stress-controlled rheometer (Discovery HR2, TA Instruments, Herts, UK) prepared with 20 mm parallel plates. For each experiment, the sample was loaded onto the stage, and the upper plate lowered until a 500 µm gap was achieved.
All readings were recorded at 37 °C. Amplitude sweeps were completed, at least one for each sample, at a fixed frequency of 1 Hz in the 0.1–10% strain range to calculate the linear viscoelastic region (LVR) of each sample.
Frequency sweeps were then completed at 1% strain, within the LVR of the samples. After, measurements of G′ and G″ were taken at 0.2% strain and frequency of 1 Hz. Repetition of all measurements was completed at least three times to guarantee reproducibility.
Live Cell Imaging: A HIV-1 derived vector consisting of a T2A peptide (EGRGSLLTCGDVEENPGP) or GFP labeling was used to transduce hdASCs and had been created to permit GFP expression and elongation factor 1α.
The lentiviral vector consisted of several cloning sites, WPRE element, SV40 polyadenylation signal, SV40 origin, pUC origin, and ampicillin resistance gene. hdASCs at passage 3 were plated in six well plates (2 × 105 cells per well) and treated with a concentration of lentivirus that has variations based on the batch being used.
Incubation of cells occurred with the lentivirus for 48 hours in standard tissue culture conditions, before the virus was removed and returned to standard tissue culture media. The GFP-labeled hdASC was used for live imaging studies because of their higher visibility of cell bodies through the thickness of the hydrogels.
Preparations of gels occurred within µ-Slide 8 Well chamber slide (Ibidi, Martinsried, Germany), and were seeded with 2 × 104 GFPlabeled hdASC. The JuLI Stage Real-Time CHR (Cell History Recorder, NanoEnTek Inc., Korea) was used for imaging and was positioned in a tissue culture incubator at 37 °C and 5% CO2.
Both attachment (first 56 hours) and proliferation (the next 45 hours) were observed for a total of 4 days. Videos were modified with NanoEnTek Inc. proprietary software.
Transmission Electron Microscopy: To evaluate the cell and gel fiber interactions, hdASC were seeded on the hydrogels at a density of 2–5 × 104 cells per insert and cultured for three days. Then, the samples were fixed with 4% formaldehyde + 2.5% glutaraldehyde in 0.1 M Hepes buffer (pH 7.2).
They were post-fixed with 1% osmium tetroxide + 1.5% potassium ferrocyanide in 0.1 M cacodylate buffer (pH 7.2) for 1 hour, before 1% tannic acid in 0.1M cacodylate buffer (pH 7.2) for 1 hour, and finally in 1% uranyl acetate in water for 1 hour.
Dehydration of the samples was completed in ethanol series infiltrated with TAAB Low Viscosity resin and polymerized for 24 hours at 60 °C. Sections were cut with Reichert Ultracut ultramicrotome and monitored with FEI Tecnai 12 Biotwin microscope at 100 kV accelerating voltage. The Gatan Orius SC1000 CCD camera was used to take images.
Analyses of Gene Expression: To evaluate the effect that SAPs hydrogels have on the hdASC phenotype, molecular analyses for key markers of stem cell differentiation toward neuroglial lineage was completed. The gene expression studies involved 1.2 × 105 hdASCs, which were seeded within each well. Six wells were plated for all substrates at each time point (days 1 and 4).
After collection, the supernatant was taken away from all gels, and 350 of RNA cell protect (QIAGEN, Manchester, UK) were added to the gels. Shaking was completed for 20 minutes at room temperature on a bench-top plate shaker at 200 rpm (MS2 minishaker, Camlab Limited, Cambridge, UK), RNA cell protect was gathered (samples were pooled in pairs, final n number n = 3) and frozen at –80 °C until RNA extraction.
The extraction of RNA was completed with the RNeasy Plus Mini Kit (QIAGEN, Manchester, UK) according to manufacturer directions. RNA concentrations were attained by spectrophotometric analyses utilizing a NanoDrop ND-100 (Thermo Fisher Scientific, Waltham, MA USA), and 400 ng of each sample was reverse transcribed utilizing RT2 First Strand Kit (QIAGEN, Manchester, UK) using manufacturer procedures.
The RNA extraction and cDNA synthesis were inclusive of DNA elimination stages to guarantee the absence of downstream genomic DNA amplification. qRT-PCR was completed using RT2 SYBR Green qPCR Mastermix (QIAGEN, Manchester, UK) using a Corbett Rotor Gene 6000 (QIAGEN, Manchester, UK) with the procedure: 10 minutes of hot start at 95 °C, followed by 40 cycles of 15 seconds at 95 °C, annealing for 30 seconds at 55 °C and finally extension for 30 seconds at 72 °C.
To confirm the specificity of reactions, a melting curve was completed with the procedure: 1 minute at 95 °C, 2 minutes at 65 °C, and a steady increase in temperature from 65 to 95 °C (2 °C min−1).
All the primers for the genes of interest and the housekeeping gene were attained from Primer Design (Chandlers Ford, UK), QIAGEN (Manchester, UK), or Sigma-Aldrich (Poole, UK), and when available the sequences are seen in Table 1.
Table 1. Primer sequences for real-time PCR studies. Source: Manchester BIOGEL
| Gene |
Forward primer |
Reverse primer |
Company |
| BDNF |
AGGTGGCTCTGGAATGACAT |
TGGGATGGTGGGCATAAGT |
Primer Design |
| NGF |
AGGAGCAAGCGGTCATCAT |
GTCTGTGGCGGTGGTCTT |
Primer Design |
| GDNF |
GCTCCAGAGACTGCTGTGTAT |
TCCTCTTCTTCTTCCTCCTCCT |
Primer Design |
| NRG-1 |
AAGTCAGAACTTCGCATTAACAAAG |
ATGGTGATATTGGCAGAGGCA |
Primer Design |
| NT-3 |
AGGTAACAACATGGATCAAAGGA |
AGGGTGCTCTGGTAATTTTCC |
Primer Design |
| TrkA |
CCCCATCCCTGACACTAACA |
GAGCAGCGTAGAAAGGAAGAG |
Primer Design |
| TrkC |
ACGAGAGGGTGACAATGCTG |
AGTTCAGATTGGTCTGGTGAGT |
Primer Design |
| Ret |
GAGACGGCTGGAGTGTGAG |
GGTGGAGAAGTTCCTGGTGAT |
Primer Design |
| Nestin |
AGAGCGTAGAGGCAGTAAAATC |
GTGCTTGAGTTTCTGGAGATTTC |
Primer Design |
| Vimentin |
TTCTCTGCCTCTTCCAAACTTT |
CGTTGATAACCTGTCCATCTCTA |
Primer Design |
| Krox-20 |
AACGGAGTGGCCGGAGAT |
ATGGGAGATCCAACGACCTCT |
Sigma-Aldrich |
| c-Jun |
GCATGAGGAAACGCATCGCTGCCT |
CCAAGT GCGACCAAGTCCTTCCCACTC GTGCACACT |
Sigma-Aldrich |
| Bax |
CCTGTGCACCAAGGTGCCGGAACT |
CCACCCTGGTCTTGGATCCAGCCC |
Sigma-Aldrich |
| BCL-2 |
GAGGTCACGGGGGCTAATT |
GAGGCTGGGCACATTTACTG |
Primer Design |
Results were normalized for the housekeeping gene (18s), and the ∆∆Ct method was used to determine the fold changes in gene expression, in comparison to Col I gels at day 1 as controls. All data were expressed as Fold Change versus day 1 Col I gels ± SEM.
Dorsal Root Ganglion Cultures and Co-Culture with hdASC: Primary cultures of DRG neurons were attained as defined previously. DRG were dissected with care from the spinal cords of adult male Sprague–Dawley rats and placed in F12 media as storage for no longer than 30 minutes.
After meticulous removal of the nerve roots, the ganglia were digested through two cycles of 0.125% (w/v) collagenase type IV (1 h each at 37 °C, Worthington Biochemical, UK), before incubating them in 0.25% (w/v) trypsin (Worthington Biochemical) for 30 minutes at 37 °C.
The blockage of enzymatic digestion occurred with the addition of fetal bovine serum, which was thoroughly rinsed with F12 medium before further mechanical dissociation with a Pasteur glass pipette. The resulting cell suspension was filtered and centrifuged for 5 minutes at 110 g; then, the cell pellet was resuspended and gradually pipetted down a gradient tail made with a solution of 30% (w/v) bovine serum albumin and F12 medium (1:1 ratio).
Following removal of the floating cell debris, the pellet consisting of DRG neurons was resuspended in Bottenstein and Sato’s (BS) medium containing 1% N2 supplement (v/v; Life Technologies, UK) and 50 ng mL NGF (Millipore, USA) in F12 medium. Finally, the neurons were plated on the variations of substrates prepared as previously described.
For the laminin-coated substrates, 10 µg µL− 1 of laminin (Sigma-Aldrich, Poole, UK) was added to the pre-set gels and the incubation period was 2 hours at 37 °C and 5% CO2; before cell seeding, laminin solution was removed, and gels were rinsed with F12 medium.
For the coculture experiments, 5 × 104 hdASC (GFP labeled as previously described for the live imaging experiments) were seeded on the substrates 24 hours before DRG collection using the stem cell differentiation media with all growth factors.
When neurons were prepared for plating, stem cell media was taken away, and DRGs were plated using a mixed medium containing a mixture of BS media and stem cell differentiation media (ratio 1:1).
For both experimental setups, cultures were fixed in 4% (w/v) PFA in PBS 72 hours after DRG plating and stained utilizing a TUJ1 specific antibody (mouse monoclonal, 1:500, SigmaAldrich) and fluorescent-labeled secondary antibodies (anti-mouse AlexaFluor-488 or anti-mouse AlexaFluor-568, both 1:1000, Thermo Scientific).
Calcium Imaging: For intracellular Ca2+ measurements, seeding of DRG neurons occurred on the top of a thin layer of hydrogels (70 µL) set on the bottom of low-adherence 24 well-plates (Corning, Tewksbury, MA, USA).
Following 72 hours incubation, cells were loaded for 40 minutes at 37 °C with 3 × 10−6 M of Fluo-4-AM (Molecular Probes, Life Technologies) in KrebsRinger-modified buffer (KRB): 136 × 10−3 M NaCl, 20 × 10−3 M HEPES, 5.5 × 10−3 M glucose, 1.2 × 10−3 M KH2PO4, 1.2 × 10−3 M MgSO4, 5 × 10−3 M NaHCO3, 1.8 × 10−3 M KCl, 2 × 10−3 M CaCl2 pH 7.4 (all from Sigma-Aldrich) supplemented with 0.01% pluronic acid (Molecular Probes, Life Technologies).
After de-esterification in KRB for 20 minutes at 37 °C, images of the DRG neurons were taken on an inverted Olympus IX-51 fluorescent microscope (Olympus, UK). Treatment of cells was completed with 50 × 10−3 M KCl to induce membrane depolarization, the generation of action potentials, and the opening of voltage-gated calcium channels.
DRG cultures were imaged for 1–3 minutes after KCl treatments. ImageJ software (NIH) was used to measure fluorescence intensities and plotted using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
Statistical Analyses: For cell viability studies, data were expressed as mean absorbance at 490 nm ± standard deviation (SD), and statistical significance was assessed by one-way analysis of variance (ANOVA) using Tukey’s multiple comparisons test.
For gene expression studies, data were expressed as fold-change of expression in comparison to the Col I control at day 1 ± SD, and statistical significance was assessed by two-way ANOVA using Tukey’s posthoc test.
For rheology experiments, data were expressed as G′ (Pa) ± SEM, and statistical significance was estimated through using a two-way ANOVA before Sidak’s multiple comparison test. All analyses were performed in GraphPad Prism (version 7.0c, Graphpad Software Inc., La Jolla, CA, US), and levels of significance were expressed as P values (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
References
[1] a) F. Thorsen, H. E. Rosberg, K. Steen Carlsson, L. B. Dahlin, J. Plast. Surg. Hand Surg. 2012, 46, 184; b) A. Faroni, S. A. Mobasseri, P. J. Kingham, A. J. Reid, Adv. Drug Delivery Rev. 2015, 82–83, 160.
[2] S. Jones, H. M. Eisenberg, X. Jia, Int. J. Mol. Sci. 2016, 17, E1494.
[3] B. He, Z. Zhu, Q. Zhu, X. Zhou, C. Zheng, P. Li, S. Zhu, X. Liu, J. Zhu, Neural Regener. Res. 2014, 9, 661.
[4] a) J. H. Bell, J. W. Haycock, Tissue Eng., Part B 2012, 18, 116; b) A. Magaz, A. Faroni, J. E. Gough, A. J. Reid, X. Li, J. J. Blaker, Adv. Healthcare Mater. 2018, 7, 1800308.
[5] A. Mobasseri, A. Faroni, B. M. Minogue, S. Downes, G. Terenghi, A. J. Reid, Tissue Eng., Part A 2015, 21, 1152.
[6] a) A. Faroni, R. J. Smith, A. J. Reid, Neural Regener. Res. 2014, 9, 1341; b) A. Faroni, G. Terenghi, A. J. Reid, Int. Rev. Neurobiol. 2013, 108, 121; c) M. Tohill, G. Terenghi, Biotechnol. Appl. Biochem. 2004, 40, 17.
[7] C. Ide, Neurosci. Res. 1996, 25, 101.
[8] R. Sullivan, T. Dailey, K. Duncan, N. Abel, C. V. Borlongan, Int. J. Mol. Sci. 2016, 17, E2101.
[9] a) A. Faroni, R. J. Smith, L. Lu, A. J. Reid, Eur. J. Neurosci. 2016, 43, 417; b) P. J. Kingham, M. K. Kolar, L. N. Novikova, L. N. Novikov, M. Wiberg, Stem Cells Dev. 2014, 23, 741; c) K. Tomita, T. Madura, Y. Sakai, K. Yano, G. Terenghi, K. Hosokawa, Neuroscience 2013, 236, 55.
[10] A. E. Mortimer, A. Faroni, M. A. Kilic, A. J. Reid, Stem Cells Int. 2017, 2017, 1.
[11] a) A. Faroni, S. W. Rothwell, A. A. Grolla, G. Terenghi, V. Magnaghi, A. Verkhratsky, Cell Death Dis. 2013, 4, e743; b) P. Erba, C. Mantovani, D. F. Kalbermatten, G. Pierer, G. Terenghi, P. J. Kingham, J. Plast. Reconstr. Aesthetic Surg. 2010, 63, e811.
[12] A. S. Hoffman, Adv. Drug Delivery Rev. 2002, 54, 3.
[13] C. M. Madl, S. C. Heilshorn, H. M. Blau, Nature 2018, 557, 335.
[14] a) F. Gelain, D. Silva, A. Caprini, F. Taraballi, A. Natalello, O. Villa, K. T. Nam, R. N. Zuckermann, S. M. Doglia, A. Vescovi, ACS Nano 2011, 5, 1845; b) M. W. Tibbitt, K. S. Anseth, Biotechnol. Bioeng. 2009, 103, 655.
[15] M. Nune, P. Kumaraswamy, U. M. Krishnan, S. Sethuraman, Curr. Protein Peptide Sci. 2013, 14, 70.
[16] a) A. Markey, V. L. Workman, I. A. Bruce, T. J. Woolford, B. Derby, A. F. Miller, S. H. Cartmell, A. Saiani, J. Pept. Sci. 2017, 23, 148; b) S. Zhang, D. M. Marini, W. Hwang, S. Santoso, Curr. Opin. Chem. Biol. 2002, 6, 865.
[17] a) J. Gao, C. Tang, M. A. Elsawy, A. M. Smith, A. F. Miller, A. Saiani, Biomacromolecules 2017, 18, 826; b) S. R. Caliari, J. A. Burdick, Nat. Methods 2016, 13, 405.
[18] a) C. Yan, D. J. Pochan, Chem. Soc. Rev. 2010, 39, 3528; b) R. V. Ulijn, A. M. Smith, Chem. Soc. Rev. 2008, 37, 664.
[19] a) K. H. Tse, M. Sun, C. Mantovani, G. Terenghi, S. Downes, P. J. Kingham, J. Biomed. Mater. Res., Part A 2010, 95A, 701; b) P. G. di Summa, P. J. Kingham, C. C. Campisi, W. Raffoul, D. F. Kalbermatten, Neurosci. Lett. 2014, 572, 26; c) P. G. di Summa, P. J. Kingham, W. Raffoul, M. Wiberg, G. Terenghi, D. F. Kalbermatten, J. Plast. Reconstr. Aesthetic Surg. 2010, 63, 1544; d) P. G. di Summa, D. F. Kalbermatten, E. Pralong, W. Raffoul, P. J. Kingham, G. Terenghi, Neuroscience 2011, 181, 278; e) K. Tomita, T. Madura, C. Mantovani, G. Terenghi, J. Neurosci. Res. 2012, 90, 1392; f) Y. Xu, L. Liu, Y. Li, C. Zhou, F. Xiong, Z. Liu, R. Gu, X. Hou, C. Zhang, Brain Res. 2008, 1239, 49; g) R. Kaewkhaw, A. M. Scutt, J. W. Haycock, Glia 2011, 59, 734; h) L. Jiang, J. K. Zhu, X. L. Liu, P. Xiang, J. Hu, W. H. Yu, NeuroReport 2008, 19, 1015.
[20] a) S. K. Walsh, R. Kumar, J. K. Grochmal, S. W. Kemp, J. Forden, R. Midha, Stem Cell Res. 2012, 8, 226; b) S. Walsh, R. Midha, Neurosurg. Focus 2009, 26, E2.
[21] J. Luo, S. Lee, D. Wu, J. Yeh, H. Ellamushi, A. P. Wheeler, G. Warnes, Y. Zhang, X. Bo, Cell Death Dis. 2013, 4, e829.
[22] a) L. Jiang, S. Jones, X. Jia, Int. J. Mol. Sci. 2017, 18; b) F. Khan, M. Tanaka, Int. J. Mol. Sci. 2018, 19, 17.
[23] a) A. Magaz, A. Faroni, J. E. Gough, A. J. Reid, X. Li, J. J. Blaker, Adv. Healthcare Mater. 2018, 7, e1800308; b) C. Kohn-Polster, D. Bhatnagar, D. J. Woloszyn, M. Richtmyer, A. Starke, A. H. Springwald, S. Franz, M. Schulz-Siegmund, H. M. Kaplan, J. Kohn, M. C. Hacker, Int. J. Mol. Sci. 2017, 18, E1104.
[24] A. M. McGrath, L. N. Novikova, L. N. Novikov, M. Wiberg, Brain Res. Bull. 2010, 83, 207.
[25] a) M. Nune, U. M. Krishnan, S. Sethuraman, Mater. Sci. Eng., C 2016, 62, 329; b) Y. Sun, W. Li, X. Wu, N. Zhang, Y. Zhang, S. Ouyang, X. Song, X. Fang, R. Seeram, W. Xue, L. He, W. Wu, ACS Appl. Mater. Interfaces 2016, 8, 2348.
[26] C. Castells-Sala, L. Recha-Sancho, A. Llucia-Valldeperas, C. SolerBotija, A. Bayes-Genis, C. E. Semino, Tissue Eng. Part C Methods 2016, 22, 113.
[27] N. Zhang, L. He, W. Wu, Neural Regener. Res. 2016, 11, 717.
[28] S. Zhang, Biotechnol. Adv. 2002, 20, 321.
[29] G. B. Schneider, A. English, M. Abraham, R. Zaharias, C. Stanford, J. Keller, Biomaterials 2004, 25, 3023.
[30] D. Kumar, V. L. Workman, M. O’Brien, J. McLaren, L. White, K. Ragunath, F. Rose, A. Saiani, J. E. Gough, Adv. Funct. Mater. 2017, 27, 1702424.
[31] a) C. M. Lo, H. B. Wang, M. Dembo, Y. L. Wang, Biophys. J. 2000, 79, 144; b) K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf, S. C. Rizzi, Biomaterials 2010, 31, 8454; c) S. R. Peyton, A. J. Putnam, J. Cell. Physiol. 2005, 204, 198; d) A. J. Engler, S. Sen, H. L. Sweeney, D. E. Discher, Cell 2006, 126, 677.
[32] R. C. Ching, M. Wiberg, P. J. Kingham, Stem Cell Res. Ther. 2018, 9, 266.
[33] a) K. R. Jessen, R. Mirsky, Glia 1991, 4, 185; b) K. R. Jessen, R. Mirsky, Nat. Rev. Neurosci. 2005, 6, 671.
[34] a) S. V. Fazal, J. A. Gomez-Sanchez, L. J. Wagstaff, N. Musner, G. Otto, M. Janz, R. Mirsky, K. R. Jessen, J. Neurosci. 2017, 37, 12297; b) K. R. Jessen, P. Arthur-Farraj, Glia 2019, 67, 421.
[35] P. J. Arthur-Farraj, M. Latouche, D. K. Wilton, S. Quintes, E. Chabrol, A. Banerjee, A. Woodhoo, B. Jenkins, M. Rahman, M. Turmaine, G. K. Wicher, R. Mitter, L. Greensmith, A. Behrens, G. Raivich, R. Mirsky, K. R. Jessen, Neuron 2012, 75, 633.
[36] R. Mirsky, K. R. Jessen, Brain Pathol. 1999, 9, 293.
[37] X. Liu, X. Wang, X. Wang, H. Ren, J. He, L. Qiao, F. Z. Cui, Acta Biomater. 2013, 9, 6798.
[38] a) N. J. Gardiner, P. Fernyhough, D. R. Tomlinson, U. Mayer, H. von der Mark, C. H. Streuli, Mol. Cell. Neurosci. 2005, 28, 229; b) N. J. Gardiner, S. Moffatt, P. Fernyhough, M. J. Humphries, C. H. Streuli, D. R. Tomlinson, Mol. Cell. Neurosci. 2007, 35, 249.
[39] J. H. Kim, Y. Park, Y. Jung, S. H. Kim, S. H. Kim, J. Tissue Eng. Regener. Med. 2017, 11, 2816.
[40] H. Yang, N. Hong, H. Liu, J. Wang, Y. Li, S. Wu, J. Cell. Physiol. 2018, 233, 9458.
[41] A. C. de Luca, C. M. Fonta, W. Raffoul, P. G. di Summa, S. P. Lacour, J. Tissue Eng. Regener. Med. 2018, 12, 676.
[42] Y. Hu, Y. Wu, Z. Gou, J. Tao, J. Zhang, Q. Liu, T. Kang, S. Jiang, S. Huang, J. He, S. Chen, Y. Du, M. Gou, Sci. Rep. 2016, 6, 32184.
[43] K. M. Koss, L. D. Unsworth, Acta Biomater. 2016, 44, 2.
[44] a) J. K. Wychowaniec, M. Iliut, M. Zhou, J. Moffat, M. A. Elsawy, W. A. Pinheiro, J. A. Hoyland, A. F. Miller, A. Vijayaraghavan, A. Saiani, Biomacromolecules 2018, 19, 2731; b) C. Ligorio, M. Zhou, J. K. Wychowaniec, X. Zhu, C. Bartlam, A. F. Miller, A. Vijayaraghavan, J. A. Hoyland, A. Saiani, Acta Biomater. 2019, 92, 92.
[45] X. Liu, A. L. Miller 2nd, S. Park, B. E. Waletzki, Z. Zhou, A. Terzic, L. Lu, ACS Appl. Mater. Interfaces 2017, 9, 14677.
[46] D. N. Heo, N. Acquah, J. Kim, S. J. Lee, N. J. Castro, L. G. Zhang, Tissue Eng., Part A 2018, 24, 537.
[47] K. A. Burgess, A. F. Miller, D. Oceandy, A. Saiani, BioTechniques 2017, 63, 253.
[48] a) W. H. Chen, S. J. Cheng, J. T. Tzen, C. M. Cheng, Y. W. Lin, PLoS One 2013, 8, e83394; b) N. D. Leipzig, M. S. Shoichet, Biomaterials 2009, 30, 6867.
[49] a) A. C. de Luca, A. Faroni, S. Downes, G. Terenghi, J. Tissue Eng. Regener. Med. 2016, 10, 647; b) A. C. de Luca, A. Faroni, A. J. Reid, J. Vis. Exp. 2015, 96, e52543.
Acknowledgments
Produced from materials originally authored by Alessandro Faroni1, Adam J. Reid1, Victoria L. Workman2 and Alberto Saiani2 from:
- Blond McIndoe Laboratories, Division of Cell Matrix Biology and Regenerative Medicine, School of Biological Sciences, Faculty of Biology Medicine and Health, University of Manchester
- School of Materials & Manchester Institute of Biotechnology, Faculty of Science and Engineering, University of Manchester
About Manchester BIOGEL

Over 15 years ago, Professors Aline Miller and Alberto Saiani at The University of Manchester synthesised a self-assembling oligo-peptide with interesting gelation properties. This work started with a small grant from the University.
Over subsequent years, the team meticulously crafted and studied self-assembling peptides to perfect their platform technology and produce a range of hydrogels ideal for 3D cell culture. In 2014, due to a demand for their materials, our company, Manchester BIOGEL was founded to enable these hydrogels to be readily available to researchers wishing to create new opportunities in the high-growth fields of 3D cell culture, 3D bioprinting and medical devices. Since opening our doors, we have supported scientists in the UK and beyond to create optimal environments for a wide variety of cell types.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.