Barrett’s esophagus requires endoscopic treatment and can often result in further damage of healthy tissue leading to fibrotic tissue formation, referred to as strictures.
This study demonstrates that synthetic, self-assembling peptide hydrogels (PeptiGelDesign) support the activity and function of primary oesophageal cells, and, in turn, epithelialization and stratification occur during in vitro 3D co-culture.
Following culture media buffering, rat oesophageal stromal fibroblasts (rOSFs) were merged into a collection of peptide hydrogels, while mouse oesophageal epithelial cells (mOECs) were planted on the surface.
Optimal hydrogels (PGD-AlphaProC and PGD-CGD2) supported the viability of mOEC (>95%), typical cell morphology, seen as cobblestone-like, and slower migration over a shorter distance in comparison to a collagen control, at 48 h.
Positive expression of typical epithelial markers (ZO-1 and cytokeratins) was found using immunocytochemistry on day 3 in culture. Optimal hydrogels were recognized, which supported rOSF viability (>95%) with homogeneous distribution when combined with the hydrogels and promoted the secretion of collagen type I found utilizing an enzyme-linked immunosorbent assay (ELISA) at day 7.
Using optimal hydrogels for both cell types, a 3D co-culture model supported a stratified epithelial layer (expressing involucrin and AE1/AE3 markers). Conclusions from this study lead to the possibility of utilizing peptide hydrogels as a minimally invasive endoscopic therapy to manage oesophageal strictures.
Introduction
Barrett’s esophagus is a precancerous condition that affects the lower esophagus. There is a presence of abnormal cells that occurs due to the metaplastic changes in the epithelium. Precisely, columnar epithelium replaces stratified squamous epithelium with the incidence of goblet cells that are mucous-producing.
The key contributing factors are believed to be directly associated with patients suffering from gastro-oesophageal reflux disease (GORD), in which recurrent exposure of stomach acid and bile salts provokes molecular changes, mutations, DNA damage, and metaplastic changes. Barrett’s esophagus is usually left untreated, though there is an increased risk of oesophageal cancer progression compared to those without Barrett’s.
Reports have shown esophageal cancer cases to be increasing in the western world. Research has reported it to be the eighth most common cancer in the world and the highest cause of cancer in Caucasian men in the UK and the USA.
Worldwide, an estimated 52,000 incidences of adenocarcinomas and 398,000 incidences of squamous cell carcinoma occur in 1 year.
Currently, the options for treatment of early oesophageal cancer include endoscopic mucosal resection (EMR) and thermal/radio ablation therapy. Both of these treatment techniques are considered safe, are commonly used, and show successful results in the removal of abnormal cells, thus reducing the risk of cancer.
However, additional damage can occur to adjacent healthy tissue, and in some cases, this causes inflammatory responses and uncontrolled fibrosis, which in turn could lead to subsequent stricture formation. This is often described as the narrowing of the esophagus.
Research shows that postoperative stricture incidences, for example, after circumferential EMR, are very high at a 70–80% risk of occurrence. Currently, stricture therapy strategies involve the use of endoscopic balloon dilation, yet, this has been associated with complications like perforation, bleeding, and bacteremia.
There is a requirement for alternative intervention to help improve current therapies, which would decrease stricture formation risk, see an improvement in patient outcomes, reduce morbidity, reduce patient revisits, and decrease costs for the healthcare systems’ through lessening the need for repeated dilation therapies and the use of dilators and stents.
Alternative strategies for the management and elimination of strictures are emerging. These strategies involve tissue engineering therapies through the use of biomaterials.
Although this area is relatively unexplored, Badylak Laboratory is the leading contributor in biomaterial-based strategy development for the engineering of oesophageal tissue and/or stricture management.
To aid with complete tissue regeneration, there have been developments in decellularized xenograft extracellular matrix (ECM, a derivative from porcine small intestine submucosa (SIS) and urinary bladder mucosa) scaffolds in the form of tubular constructs. Preclinical research has shown endoscopic surgery to be successful in placing these tubular constructs over the damaged site in canine models.
Results found that after eight weeks, there was esophageal mucosal remodeling with complete re-epithelialization and no stricture formation. SIS ECM scaffolds have also been implanted into five human patients.
This study involved ECM scaffolds being retained in place with an Ultraflex stent, which was then removed between 9 and 18 days after implantation.
Results showed minimal stricture formation and by four months, and almost complete mature esophageal squamous epithelialization was seen. More recent advances in this area have involved the use of hydrogels. These have already shown promises for numerous areas of regenerative medicine and tissue engineering applications.
However, these advances have only just been investigated for gastrointestinal (GI) tract applications, specifically for esophageal stricture management. In comparison to decellularized tubular scaffolds, hydrogels have several benefits.
For example, these benefits include injectability, the use of minimally invasive procedures, room for repeat administrations, modified mechanical properties, the capabilities to fill an irregularly shaped space, and bioactivity to mimic native tissue ECM.
For the reasons mentioned above, the development of decellularized ECM hydrogels has progressed, also providing an opportunity for stricture management in the future. Although oesophageal stricture management using hydrogels is a reasonably unexplored area, recently, several studies have been performed to aid with this progression.
For instance, as combined therapy with balloon dilation, a steroid-loaded hydrogel, named Endolubri jelly, was sprayed endoscopically into 20 patients. Early results from these studies showed promising outcomes where a complete squamous epithelium without stenosis was seen after three months.
Further developments involve the use of the Food and Drug Administration (FDA)-approved hydrogels for early treatment and enhanced prognosis of Barrett’s esophagus with specific attention paid to the lower oesophageal sphincter (LOS). When the LOS weakens in patients, it can lead to the development of GORD.
A variety of mucoadhesive hydrogels, specifically, Polaxamer 407, cross-linked polyacrylic acid, hydroxypropylmethylcellulose, sodium carboxymethylcellulose, and chitosan were used in one study and provided a transport material for the topical delivery of hexylaminolevulinate (HAL), an optical imaging agent. This was completed in an ex vivo rat model and four healthy human males.
It was revealed that for esophageal adhesion, chitosan was the optimal hydrogel in vitro and in vivo. Additionally, in vitro release profiles from chitosan loaded with 40 × 10−3 M of HAL showed that after a residence time of 10 minutes on the wall of the esophagus, the amount of HAL transported to the epithelium was adequate to allow fluorescence diagnosis of Barrett’s esophagus.
In vivo studies established that increased concentrations of chitosan (Protasan) from 1.5% to 1.7% enhanced adhesivity and the fluorescence signal, with a full-thickness layer coating on the mucosa.
The remit of the application was completely focused on improving prognosis. However, Johnson et al. conducted a study that focused on LOS improvement.
During the research, a nonresorbable polymer, ethylene vinyl alcohol, was injected into the LOS via a sclerotherapy-type needle. When being injected, the polymer acts as a nonviscous liquid, which quickly solidifies in situ.
This medical device, called Enteryx, was made commercially available, but recent reports lead to the removal of the device from the market by the FDA due to patient injury and death.
Alternative types of hydrogel, decellularized ECM hydrogels (a derivative from porcine dermal and urinary bladder matrix (UBM)) have recently been developed and have shown promising results.
Studies took place in a rat abdominal wall defect model, and after in vivo implantation, both hydrogels had degraded by 35 d, and UBM hydrogels were proven to support higher amounts of myogenesis in comparison to dermal hydrogels.
Progressions towards clinical trials have been recognized in decellularized hydrogels and are currently being examined for oesophageal strictures for the facilitation of repair and reconstruction.
Hydrogel alternatives made of ECM/biological materials are those created using de novo designed materials. Great interest has been shown in the progression and use of synthetic, self-assembling peptide hydrogels in the past decade.
A range of designs of short peptides has been offered. These designs self-assemble into water-swollen networks (hydrogels) above a critical gelation concentration (CGC). A study by Zhang and colleagues formulated a family of amphipathic self-assembling peptides based on the alternation of hydrophilic and hydrophobic residues.
These peptides showed capabilities to self-assemble into β-sheet-rich nanofibrillar structures, usually explained as 3 nm fibers and 10-50 nm bundles. These above the CGC, entangle and/or associate with form hydrogels.
The propensity of these peptides to self-assemble and the nature of the nano-structures and general properties of the hydrogel formed depend on many factors. These include the amino acid sequence, the peptide concentration to start, the pH, and the type of electrolyte in the medium.
Peptide hydrogels are now commercially available from many businesses, such as Puramatrix (Corning), HydroMatrix (Sigma), Biogelx, and, more recently, PGD-HydroGels (PeptiGelDesign).
There are numerous benefits in using synthetic, self-assembling peptide hydrogels, like their low cost, consistency, controllable manufacturing, the fact that amino acids can be simply metabolized by enzymes in vivo, leading to a highly biocompatible gel, and that they are endoscopically deliverable via spraying or injection.
A wide range of properties is offered through altering both the amino acids present and their order within the peptide sequence.
The existence of charged amino acids permits variation in both net charge and charge distribution. Using this system, tailored release properties, rates of degradation, and mechanical properties are all possible and are hard to achieve with natural polymers.
This article suggests using self-assembling peptide hydrogels for a new application to treat and/or manage strictures caused by ER for the removal of metaplastic cells in Barrett’s esophagus.
It can be predicted that after delivering acellular self-assembling peptide hydrogels to the injured area, the healthy adjacent tissue cells would have capabilities to migrate, yield essential matrix proteins, and allow tissue regeneration.
Delivering the hydrogel endoscopically is feasible due to the shear-thinning and self-assembling properties of these hydrogels.
After the hydrogel has been delivered to the injury site, it can be expected that host fibroblasts and epithelial cells will migrate into the hydrogel from adjacent healthy tissue, allow re-epithelialization and production of a matrix, and allow normal function in acting as a protective, semipermeable barrier.
Swift restitution of the mucosal barrier would lead to a decreased inflammatory response resulting in successive modulation of fibrosis.
Overall, the production and remodeling of the neo-tissue matrix would occur with the chosen native tissue-type matrix composition and mechanical properties. To progress towards this final translational goal, initial studies took place in vitro to assist the understanding of tissue-specific (stromal fibroblasts and oesophageal epithelial) cell response to a library of synthetic peptide hydrogels.
Due to different cells needing distinct extracellular matrix conditions, researchers in this study speculate that each cell type needs a specific gel property. PeptiGelDesign can offer a family of related peptide hydrogels, each with specific charge and mechanical property combinations.
Utilizing the panel allowed for evaluations of oesophageal cell response and cell function characterization through morphology, migration, viability, and typical marker expression. This allowed the optimal recognition of hydrogels for each cell type.
Additionally, this showed the initial development of a 3D co-culture, a composite model using self-assembling peptide hydrogels in order to show the multilayer and multicellular nature of oesophageal submucosa.
The unique properties of this hydrogel system permitted gel combinations to be layered, providing distinct conditions appropriate for supporting each cell type.
This study delivers the first steps for progressing in using synthetic peptide hydrogels in oesophageal stricture management and the potential for oesophageal tissue engineering.
Results
Rheological assessment of peptide hydrogels
After buffering in media for 24 hours at 37 °C, the stiffness of peptide hydrogels was evaluated. The initial pH of each hydrogel differed, as seen in Table 1, and hence the buffering step in media was vital for the diffusion of nutrients and molecules into the gels as well as neutralizing the pH for a cell-friendly environment.
Statistical analysis revealed significant stiffness (G′) in PGD-Alpha1, PGD-Alpha2, PGD-AlphaProC, and PGD-CGD2 hydrogels in comparison to PGD-AlphaProB and PGD-C2 hydrogels (p < 0.001; Table 1).
Table 1. Peptide hydrogels mechanical properties and charge density supplied by PeptiGelDesign. Source: Manchester BIOGEL
| Hydrogel |
Stiffness G' after media
conditioning for 24 h [Pa] |
Initial pH |
Charge density |
| PGD-Alpha1 |
15 000 ± 5000 |
3.0–3.5 |
Neutral |
| PGD-Alpha2 |
21 000 ± 1000 |
4.2–4.8 |
Low |
| PGD-AlphaProC |
14 000 ± 1300 |
6.9–7.1 |
High |
| PGD-CGD2 |
5500 ± 1700 |
6.0–7.0 |
Low |
| PGD-AlphaProB |
500 ± 80 |
6.3–6.7 |
High |
| PGD-C2 |
400 ± 10 |
6.0–6.6 |
High |
Mouse oesophageal epithelial cell (mOEC) response to peptide hydrogels in 2D culture
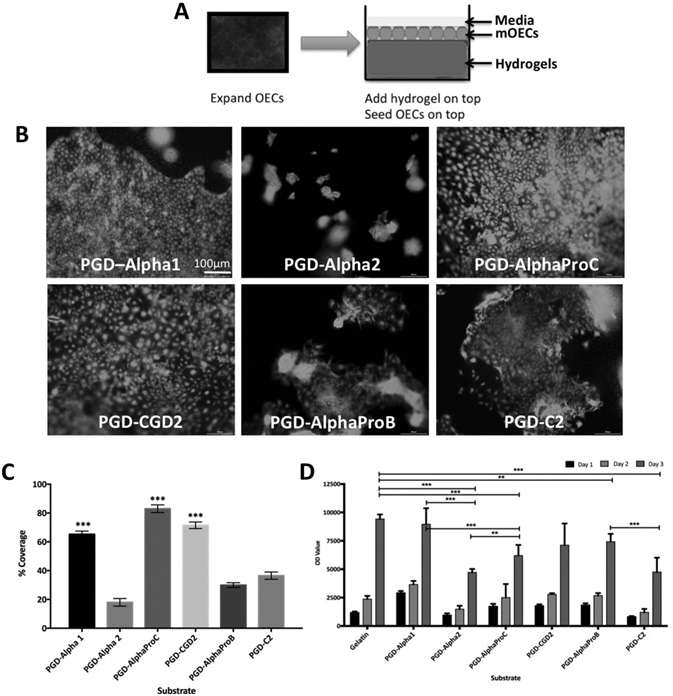
Figure 1. mOEC behavior when cultured on the surface of peptide hydrogels up to day 3. A) Schematic of experimental setup. B) Viability assessment using Live/Dead assay at day 3. C) Metabolic activity of mOEC assessment using alamarBlue assay up to day 3. D) Epithelial coverage on surface of peptide hydrogels at day 3. Values indicate average viability (D) or average percentage epithelial coverage as determined by image analysis in part (C), ±SD where n = 3. Image Credit: Manchester BIOGEL
As demonstrated in Figure 1A, mOECs were cultured on top of peptide hydrogels for 3 days and submerged in media. Live/Dead assays were used for a viability assessment and showed that mOECs responded in a different way depending on which peptide hydrogel surface they were cultured.
However, at the end of 3 days of culture, mOECs were shown to be viable on all peptide hydrogels, as seen in Figure 1B. Apparent effects on the morphology of mOEC and on the ability to form an epithelial sheet were seen as a response to the type of peptide hydrogel surface.
Results revealed that PGD-Alpha1, PGD-AlphaProC, and PGD-CGD2 supported characteristic mOEC morphology and epithelial sheet formation, whereas the epithelial sheet was not as obvious on PGD-Alpha2, PGDAlphaProB, and PGD-C2 hydrogels, and the mOECs seemed to clump together. This is displayed in Figure 1B.
Staining images were used to semi-quantify epithelial cell coverage using an ImageJ plug-in Live/Dead. These data further confirmed that PGD-Alpha1, PGD-AlphaProC, and PGD-CGD2 supported the best epithelial sheet coverage with 81%, 77%, and 65% mOEC coverage on respective peptide hydrogels. This is displayed in Figure 1C.
The alamarBlue assay was used to evaluate metabolic activity and proliferation activity of mOECs cultured on top of peptide hydrogels. All peptide hydrogels, including the control, a gelatin-coated tissue culture plastic (TCP), supported a rise in metabolic activity and proliferation from day 1 to day 3.
After 3 days of mOEC culture on top of the peptides, cells cultured on PGD-Alpha1, PGD-CGD2, PGD-AlphaProB, and PGD-C2 exhibited comparable metabolic activity to the control with differences seen to be insignificant, as seen in Figure 1D.
However, although they supported comparable metabolic activity, hydrogels PGD-Alpha2, PGD-AlphaProB, and PGD-C2 did not support an intact epithelial cell sheet formation, as displayed in Figure 1B.
Ideal peptide hydrogels, PGD-Alpha1, PGD-AlphaProC, and PGD-CGD2, as acknowledged from the above data, were explored further to characterize the expression of typical epithelial markers by mOECs.
Results showed ZO-1 staining in mOECs cultured on optimal peptide hydrogels at day 3, with similar expression to the gelatin control, as shown in Figure 2. Positive expression of a multipanel of cytokeratins secreted by mOECs was also seen when cultured on optimal peptide hydrogels on day 3.
Similar expression patterns were revealed as the control, as seen in Figure 2, further confirming comparable mOEC behavior. PGD-AlphaProC and PGD-CGD2 hydrogels supported better expression of ZO-1 and cytokeratins in comparison to PGD-Alpha1 and were advanced to a migration assay.
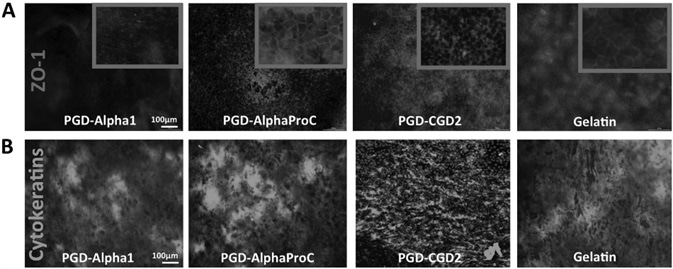
Figure 2. Characterization of typical epithelial markers. A) ZO-1 marker and B) multipanel of cytokeratins expression by mOECs cultured on the surface of optimal peptide hydrogels at day 3. Scale bar = 100 μm. Image Credit: Manchester BIOGEL
mOEC migration activity across peptide hydrogels during 2D culture
Investigations were completed to explore the ability of mOECs to migrate across candidate peptide hydrogels. They were done using a ring migration assay and time-lapse imaging over a 48-hour period, as seen in Figure 3A.
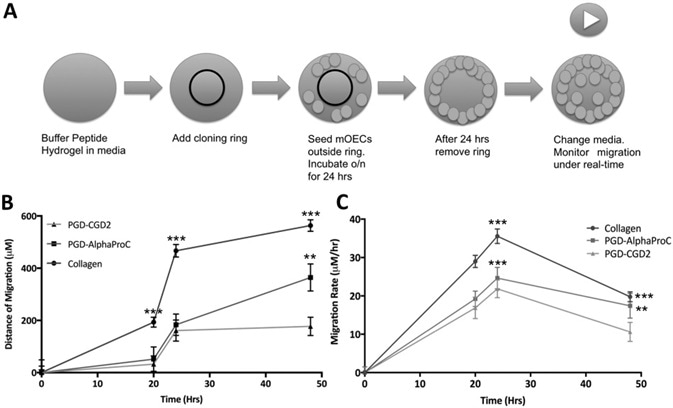
Figure 3. Determination of the migration activity of mOECs across the surface of optimal peptide hydrogels cultured up to 72 h. A) Schematic of experimental setup, B) migration distance, and C) migration rate. Values indicate the average migration distance (μm) or migration rate (μm h−1) and ±SD where n = 3. Image Credit: Manchester BIOGEL
Pixels were then translated into distance covered using ImageJ. Results showed that migration of mOECs was accomplished on both control (collagen hydrogels) and peptide hydrogels (PGD-AlphaProC and PGD-CGD2) over 48-hours.
A significantly greater mOEC distance covered was seen on collagen hydrogels in comparison to peptide hydrogels at 20, 24, and 28 hours (all time points). This is displayed in Figure 3B. Yet, a significantly greater distance was covered on PGD-AlphaProC at 364.4 µm, compared to PGDCGD2 a t177.1 µm at 48 hours.
Migration rate analysis showed a similar trend to migration distance, as shown in Figure 3C. As expected, significantly faster migration rates were seen on collagen hydrogels in comparison to peptide hydrogels at all time points (20, 24, and 48 hours).
Yet, a significantly faster migration rate was seen on PGD-AlphaProC at 17.432 µm h−1, in comparison to PGD-CGD2 at 10.625 µm h−1 at 48 hours. The migration of epithelial cells was supported by both hydrogels and so, in combination with the data shown previously, were considered appropriate for culturing mOEC.
Rat oesophageal stromal fibroblast (rOSF) response to peptide hydrogels in 3D culture
rOSFs were merged into a panel of buffered, peptide hydrogels and cultured up to 7 and 14 days in media, as demonstrated in Figure 4A. Live/Dead staining showed that rOSFs incorporated into all peptide hydrogels and remained viable after 7 and 14 days of culture (Figure 4B). Differences in rOSF morphology were also obvious on day 14, depending on which peptide hydrogels in which they were cultured.
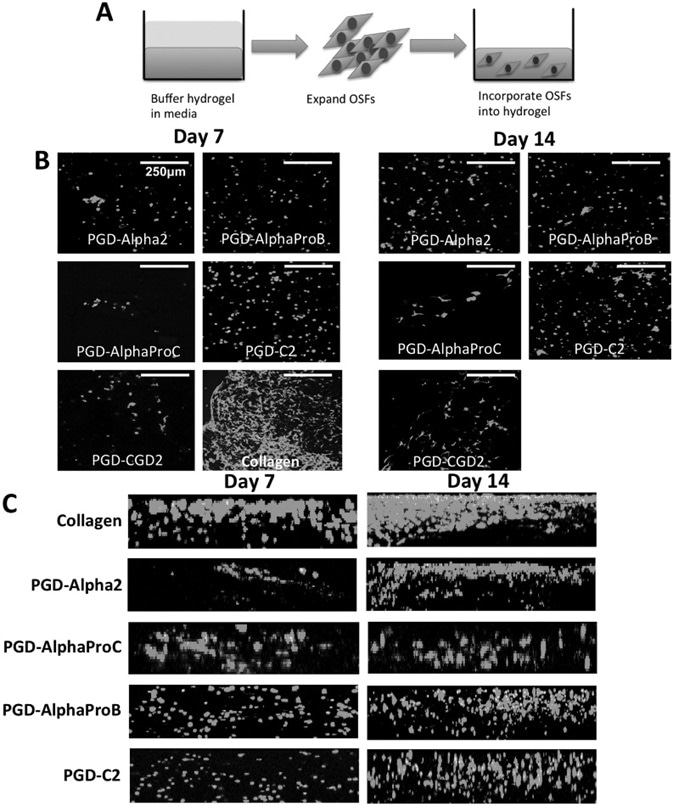
Figure 4. The behavior of rOSFs embedded within a panel of peptide hydrogels for 3D culture up to day 14. A) Schematic of experimental set-up. B) Assessment of rOSF viability and morphology using Live/Dead assay. C) Assessment of rOSF distribution within the hydrogels using Live/Dead assay. Scale bar = 100 μm. Image Credit: Manchester BIOGEL
rOSFs were deemed to be more fibroblastic in PGD-AlphaProC, PGD-C2, and PGD-CGD2 hydrogels, but remained grounded in PGD-Alpha2 and PGD-AlphaProB, rOSFs, at day 14.
As anticipated, rOSFs cultured in collagen hydrogels kept their typical fibroblastic-like morphology, and more cells were present on both days 7 and 14 in comparison to all peptide hydrogels that were explored.
The homogeneous distribution of the cells throughout the hydrogel was determined through confocal microscopy. Observing the distribution of rOSFs in the peptide hydrogels gave way to establishing whether the hydrogel environment supported cell growth and migration, or if cells gathered together in a survival attempt.
Using confocal microscopy, figure 4C signifies the full thickness (≈200 µm) view of z-stack Live/Dead images that were compiled.
As anticipated, a larger number of rOSFs were seen in collagen hydrogels at days 7 and 14 when compared to the peptide hydrogels, as seen in Figure 4C. rOSFs were seen to be most homogeneously distributed when encapsulated in PGD-AlphaProB and PGD-C2 at both time points, followed by collagen hydrogels (Figure 4C).
Table 1 shows both of these gels to be mechanically weaker. On the contrary, rOSFs looked to group together, and heterogeneously distribute themselves within PGD-Alpha2 and PGD-AlphaProC at both time points (days 7 and 14). Both of these gels are seen to be the stiffest gels in the panel, as seen in Table 1.
Immunohistochemistry and quantification of collagen type I using ELISA
Immunohistochemistry was used to determine the type I collagen secretion into the hydrogels by rOSFs. Observations showed a higher number of rOSFs present in PGD-C2 in comparison to PGD-AlphaProC at both time points on days 7 and 14.
In addition to this, higher amounts of type I collagen were seen surrounding individual rOSFs in PGD-C2 in comparison to PGD-AlphaProC, as demonstrated in Figure 5A.
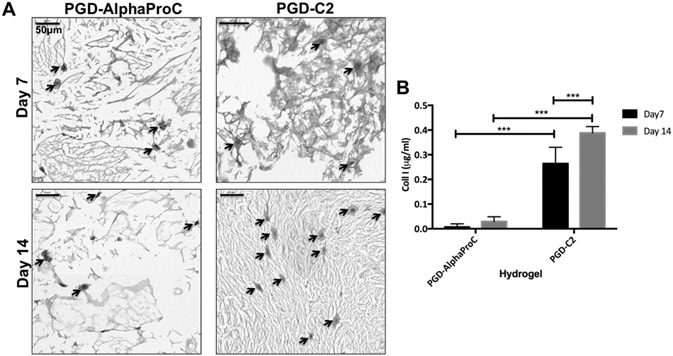
Figure 5. Characterization of type I collagen production. A) Immunohistological images of type I collagen production by rOSFs cultured in PGDAlphaProC and PGD-C2 hydrogels after 7 and 14 d. B) Quantification of type I collagen production by rOSFs cultured in PGD-AlphaProC and PGD-C2 hydrogels at days 7 and 14. Values indicate mean amount of collagen values ±SD where n = 3 (three replicates in a single experiment). Arrows indicate the presence of rOSFs surrounded by deposition of collagen type I within the hydrogels. Image Credit: Manchester BIOGEL
For further results to determine and quantify the secretion of type I collagen into the hydrogel (PGD-AlphaProC and PGD-C2), ELISA was used to quantify the collagen in constructs cultured up to days 7 and 14.
A rise in collagen production from day 7 to day 14 was revealed in both hydrogels, PGD-AlphaProC (0.007–0.0295 µg mL−1 ), and PGD-C2 (0.264–0.3884 µg mL−1). PGD-C2 showed the greatest production of type I collagen at days 7 and 14 in comparison to PGD-AlphaProC. This is shown in Figure 5B.
Considering all data, PGD-C2 was shown to be the superior peptide hydrogel for supporting rOSF growth, homogeneous distribution, and collagen type I secretion. PGD-AlphaProC was also selected for additional examination, as cells grown in this peptide hydrogel appeared to be more fibroblastic in morphology.
There was also interest in determining if a single peptide hydrogel would be appropriate for growing both cell types, which would result in a simplification of clinical translation; this was another reason for choosing PGD-AlphaProC.
Histological evaluation of the 3D co-culture composite model
Ideal performing peptide hydrogels for the 2D culture of epithelial cells (PGD-AlphaProC) and 3D culture of fibroblast cells (PGD-C2) were progressed onwards to create composite hydrogel systems in order to evaluate 3D co-culture.
Alternative systems were created. Specifically, system 1 (PGD-AlphaProC for both cell types) and system 2 (PGD-C2 incorporating OSFs; PGD-AlphaProC with OECs on top) were created and were cultured up to day 7. This is shown in Figure 6.
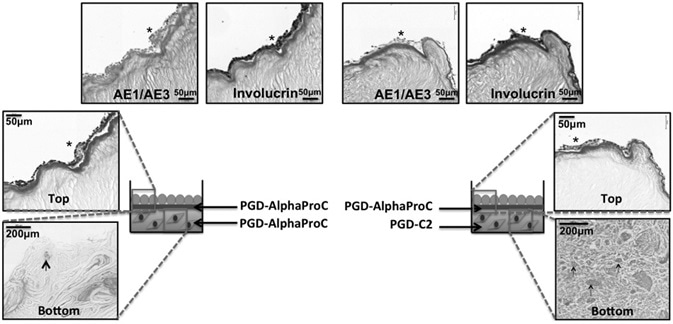
Figure 6. Histological analysis of 3D co-culture composite systems, cultured up to day 14 at air–liquid interface. Schematic of the two co-culture systems (system 1 and system 2) with H&E staining, AE1/AE3 multicytokeratin staining, and involucrin staining. Asterisk indicates the positive expression of specific epithelial markers (AE1/AE3 or involucrin), and the arrows are pointing to single rOSFs with surrounding collagen deposition. Image Credit: Manchester BIOGEL
For both systems, a successful and uninterrupted layer of mOECs was created at day 7 with a typical multicellular layer as seen using histological evaluation using hematoxylin and eosin (H&E) staining.
Histological staining for specific epithelial markers showed that there was a positive expression of AE1/AE3 and involucrin for both systems on day 7, as seen in Figure 6. Additionally, rOSFs were seen within the bottom hydrogel component of both composite systems, where the rOSFs exhibited a rounded morphology.
Discussion
This is the first study to report the response of primary oesophageal cells (epithelial cells and stromal fibroblasts) to synthetic self-assembling peptide hydrogels. A library of peptide hydrogels, as seen in Table 1, was investigated, with each showing unique physical characteristics that included stiffness and overall net charge.
The nature of this study was complex, and so it was clear that an accessible material with mechanical properties that are easy to tailor would be advantageous to support each cell type. Self-assembling peptides are markedly tailorable, and hence were the main candidate for the remit of this study.
The rationale behind using peptide hydrogels was to find a synthetic matrix with the ability to support mucosal regeneration as a first step toward the development of a tissue-engineered therapy for stricture formation management following endoscopic treatment for Barrett’s esophagus.
The results of this study clearly show that the behavior (morphology, proliferation, and intact epithelial cell sheet formation) of mOECs was influenced by the properties of peptide hydrogels, despite the fact it demonstrated similar cell viability across the panel.
Having a library of a variety of peptide hydrogels permitted the recognition of the optimal gel to be selected for each cell type mOEC and rOSF.
The performance of such assays has taken place in other studies with a similar remit where it was necessary and specifically highlighted to validate tissue-specific cell behavior and functionality in vitro prior to moving toward in vivo.
After 3 days of mOEC culture on top of buffered peptide hydrogels, the preferential peptide hydrogels were found to be PGD-AlphaProC and PGD-CGD2, as established by the characteristic cobblestone-like morphology and tight junction formation between cells (ZO-1 expression), leading to an epithelial sheet-like formation.
Observations also showed a positive expression of cytokeratins. Remarkably, the stiffest hydrogels in the panel were PGD-AlphaProC and PGD-CGD2, ranging between 5.5 and 14 kPa.
In order to signify the mucosa of the esophagus, a structure that can be easily damaged during oesophageal surgery, primary stromal fibroblasts were incorporated and cultured in 3D in buffered peptide hydrogels.
As seen previously, behavior differences were observed in terms of cell morphology, and homogeneous distribution was seen and influenced by the peptide hydrogel properties.
The optimal hydrogels were discovered to be PGD-AlphaProC, noted as the third stiffest in the panel, and PGD-C2, the weakest in the panel at 0.4 kPa. This was based on the cells’ ability to remain viable and to adopt a fibroblastic phenotype.
The difference in such cell behavior can be associated with several different material-related factors. Certainly, each factor will have a combinatorial effect on cell response. However, for precision, each factor will be examined separately.
The panel of peptides applied in this study has differing numbers of charged amino acids present, thus giving variation to the charge of the assembled hydrogels. The overall charge carried on hydrogels can have a significant influence on cell adhesion and behavior.
Commonly, positively charged surfaces increase cell attachment and proliferation as shown by studies investigated by Schneider et al. Negatively-charged hydrogels have also demonstrated increases in proliferation of endothelial cells and improvements to cartilage regeneration in vivo.
However, decreased fibroblast proliferation has been observed by Liu et al. on negatively charged hydrogels.
These variations in results regarding which charge is favored by cells to lead to the speculation of certain cells preferring a particular charge, whether that is positive or negative.
However, the current study indicates that the charge effect is dominating the effect of mechanical properties. The mechanical properties, for instance, of PGD-Alpha1 (15 kPa) and PGD-AlphaProC (14 kPa) are extremely similar, whereas the charges are neutral and highly positive, respectively.
Even though the mechanical properties are extremely similar, both cell types had different responses to the two hydrogels. Neither of the cell types chose PGD-Alpha1, suggesting a preference for positively charged material over neutrally charged.
There are undeniably synergistic responses between mechanical properties, charge, and other factors, and charge cannot be the only factor responsible for the reason cells prefer particular gels.
Cell response can also be impacted by the differences in mechanical properties of the self-assembled hydrogels used in this study. Differential rates of proliferation and cell spreading have been extensively reported within hydrogels of varying stiffness, as reviewed in Ahearne’s research.
Additionally, matrix stiffness has been shown to be in command of variations in cell phenotype, proliferation, cytoskeleton, and mobility, as well as driving stem cell differentiation toward specific lineages.
Growing both cell types with the panel of hydrogels with varying mechanical properties showed that mOECs were proven to have a preference for stiffer peptide hydrogels when compared to rOSFs, which, when encapsulated in three dimensions need weak gels for motility.
Additional analysis showed that there is a fine line in the preferred stiffness (between 5.5 and 14 kPa) of the hydrogels for mOEC attachment and expansion.
For the softer peptides (PGD-AlphaProB and PGD-C2), mOECs did not have the ability to keep a flat epithelial sheet and, instead, disconnected from the hydrogel, demonstrating weak attachment and interaction of mOECs to the surface of these hydrogels.
Similarly, on PGD-Alpha2, the stiffest hydrogel in the panel, cells were unable to attach. There is a need for further studies to profile cell surface integrin binding and cytoskeleton behavior to clarify how well mOECs anchor and/or interrelate with the substrate and whether interaction changes occur as a result in stiffness change.
Hydrogel stiffness also had an influence on rOSF behavior. Specifically, after encapsulation, the homogeneous distribution within the hydrogels was improved within the softer hydrogels (PGD-AlphaProB and PGD-C2).
Remarkably, the rise in cell number seemed to remain constant across the panel of hydrogels, as seen as a visual example in the Live/Dead images from day 7 to day 14. This possibly suggests that the hydrogels may have an intrinsic property to modulate fibroblastic proliferation activity.
However, further investigation is needed on this property. It appears that there is a conclusive combination of material-related factors (charge and mechanical properties) that operate in collaboration to influence the cell response effect, of which some are yet to be completely defined and understood.
Additional advantages of these peptide hydrogels are that they have a nanofibrillar architectural structure, similar to that of native ECM, and are shear thinning. This property provides the opportunity to transport/inject the hydrogel material, either in acellular form or as a cell delivery vehicle, in a minimally invasive technique through endoscopy.
The requirements of this delivery include minimal deviation from currently used oesophageal treatments, which are performed endoscopically. In addition, the peptide hydrogels that mOECs responded well to (PGD-AlphaProC and PGD-CGD2) also have mechanical properties similar to the native human esophagus tissue interface (5–50 KPa).
Interestingly, after media conditioning, most of the hydrogels on the panel had G′ values above 5 kPa, with the exceptions of PGD-AlphaProB and PGD-C2.
Although this study was the primary stage in the direction of the development of a tissue engineering therapy, further future investigations are needed to explain the degradation profile of the optimal hydrogels prior to progression towards in vivo implantation studies.
These degradation studies are required to confirm that the gel did not undergo degradation or lose mechanical stiffness within the time period needed for epithelialization.
Utilizing hydrogel, studies could anticipate the promotion of early epithelialization, which would later minimize related proinflammatory responses thought to be vital in preventing stricture formation.
Additional in vitro and in vivo studies are also needed to determine the ability of the hydrogel to modulate inflammatory responses and the formation of fibrotic tissue.
As this study showed a positive oesophageal cell response to the known peptide hydrogels in vitro, alternative strategies involving cell-loaded hydrogels could be the delivery of an acellular peptide hydrogel in vivo.
Migration data supported that mOECs could migrate across the surface of the peptide hydrogel and allow epithelialization. By imitating this situation in vivo post-implantation, the epithelial cell migration from healthy adjacent tissue on to the peptide hydrogel and the hydrogel to competently allow re-epithelialization of the esophagus can be envisaged.
Further, an acellular product shows an increasingly possible route to market and one with reduced risks and apprehensions due to the absence of biological material.
This is the initial study that has tried to replicate the in vivo oesophageal mucosa structure (epithelium and stroma, which are typically damaged during endoscopic surgery and treatment) of the esophagus in vitro by evaluating cell response when in 3D co-culture within synthetic peptide hydrogels.
Other in vitro 3D co-culture systems have occurred, but these have been developed using xenogenic material. For this study, a composite hydrogel system was created as the two different cell types in focus (mOECs and rOSFs) grew favorably on different hydrogels.
The top layer supported the epithelial layer and was cultured at the air-liquid interface to mimic the environment in vivo of the oesophageal epithelium.
Both composite systems histological analysis (cultured for 7 days) showed the existence of stromal fibroblasts within the bottom layer hydrogel and a distinct, continuous epithelium layer on the surface of the top-layered hydrogel.
On both composites, there were 2-3 layers of cell in the epithelium, therefore suggesting that stratification of the epithelium began to take place after 7 days of culture. The cell layer amount can be attributed to the crosstalk between stromal fibroblasts and epithelial cells when in co-culture.
In a study similar to this one, an organotypic model with a combination of xenogenic material containing collagen hydrogel and Matrigel with incorporated stromal fibroblasts was cultured for 7 days prior to seeding oesophageal epithelial cells on top and continuing culture up to day 15.
Although there was a contraction of the hydrogel by the stromal fibroblasts, the crosstalk between both cell types encouraged a stratified epithelium to be created on top with the positive expression of involucrin and cytokeratins.
In the current study, the hydrogels were not visually contracted by the stromal fibroblasts, but still supported the creation of epithelium within an organotypic (co-culture) model.
This highlighted the difference in fibroblast morphology, which is dependent on the culture conditions. For instance, a fibroblastic morphology was seen when fibroblasts were cultured in 3D, whereas under co-culture conditions, fibroblasts remained rounded.
Commonly, it is accepted that communication occurs between the epithelial cells and fibroblasts in the submucosa via paracrine signals, which influence cell differentiation, proliferation, and function.
It is hypothesized in this study that this signaling phenomenon may be accountable for the differences in morphology of fibroblasts that have been observed.
It is important for the biomaterial of choice to promote stratified epithelialization as this specifies a thick epithelium with suitable function, like creating a protective barrier. Only a small number of studies have tried to gain a better understanding of the interaction of epithelial cells to appropriate substrates in vitro.
Beckstead et al. examined oesophageal epithelial behavior on an array of synthetic scaffolds (polyesters) in addition to natural (Alloderm, defined as decellularized skin graft).
Alloderm supported the creation of a 5–6 cell layer epithelium with the expression of loricrin, a late-stage marker for epithelium (similar to involucrin), after 18 days in culture.
Conversely, the synthetic scaffolds only supported an epithelium layer of 2–3 cells thick, which lacked overall spatial organization and created an interrupted layer. This lack of epithelialization could be due to the pore size, structure, and surface properties of the scaffolds.
Used in the current study, synthetically-produced peptide hydrogels also supported an epithelium that was 2–3 cells thick. However, in this case, it created an uninterrupted layer within an enhanced time period of 7 days, and also expressed a late-stage differentiation marker (involucrin).
The outcomes observed show that the structural and surface properties, in addition to the pore size, maybe architecturally suitable for supporting the creation of a functional epithelium.
Further to this, synthetic peptide hydrogels may eliminate the requirement of pre-coating synthetic scaffolds with ECM proteins like collagen and fibronectin to support the expansion of oesophageal epithelial cells. This provides a minimally invasive therapy, which is reproducible and has the potential to be mass-produced.
Conclusion
Synthetic peptide hydrogels, PGD-AlphaProC and PGD-CGD2, have been recognized to support mOEC adhesion, typical morphology, proliferation, and epithelial cell sheet formation with positive expression of typical markers (AE1/AE3 and involucrin).
Additionally, synthetic peptide hydrogels, PGD-AlphaProC, and PGD-C2, were seen to support the 3D homogeneous distribution of viable rOSFs. A co-culture model was set up consisting of both cell types, mOECs, and rOSFs, representing a multicellular and multilayer hydrogel system.
Successful support of the co-culture system was seen in the formation of a functional, uninterrupted epithelial sheet within 7 days of culture.
This article highlights the preferential synthetic peptide hydrogels to support cytocompatibility as opposed to oesophageal cells and is the first stage in identifying suitable materials or development of a tissue engineering therapy as a post-endoscopic treatment for stricture management in Barrett’s esophagus.
Experimental section
Peptide Hydrogels: A library of synthetic, self-assembling peptide hydrogels was acquired from PeptiGelDesign, located in Cheshire, UK. Each hydrogel is different in relation to the peptide sequence, stiffness, and overall charge that the peptide holds.
PeptiGelDesign used γ irradiation to sterilize the peptide hydrogels prior to cell culture, and rheological properties of hydrogel samples were taken after conditioning with media for 24 hours at 37 °C in a humidified CO2 incubator.
All rheological studies were done utilizing a stress-controlled rheometer (Discovery HR2, TA Instruments, Herts, UK) with 20 mm parallel plates.
During each experiment, 200 µL of the sample was added onto the stage, and the upper plate was lowered until it reached a 500 µm gap. All readings were recorded at 37 °C. For each sample, amplitude sweeps were performed at least once at a fixed frequency of 1 Hz in the 0.1–10% strain range in order to determine the linear viscoelastic region (LVR) of each sample. After, frequency sweeps were performed at 1% strain, within the LVR of the samples.
Subsequently, measurements of G′ and G″ were taken at 0.2% strain and frequency of 1 Hz. Repetitions of all measurements were recorded at least three times to guarantee reproducibility.
Oesophageal Cell Expansion: CellBiologics (catalog #: C57-6046, USA) provided the primary mouse oesophageal epithelial cells, and they were cultured as instructions showed in the manufacturers’ protocol.
Flasks were temporarily pre-coated with 0.2% gelatin (30 minutes at room temperature (RT)), washed twice with phosphate-buffered saline (PBS), and seeded with OECs.
Cells were cultured in epithelial cell medium and supplement kit (catalog #M6621-kit; Caltag, UK), which contained basal media supplemented with 2 × 10−3 M l-glutamine (LG), antibiotic antimycotic solution (A&A; 100 U penicillin, 0.1 µg streptomycin, and 0.25 µg mL−1 amphotericin), 5% fetal bovine serum (FBS), insulin–transferrin–sodium selenite, hydrocortisone, and epidermal growth factor.
Change in media occurred every other day, and cells were passaged at approximately 70% confluence using trypsin (0.5 g; Sigma, T3924) and reseeded at a split ratio of 1:3.
Primary rat oesophageal stromal fibroblasts were kept completely separate from Sprague Dawley rat oesophageal stromal tissue donated as waste tissue from an additional animal study.
After sacrifice, esophagus tissue was washed for 5 minutes in PBS with A&A (100 U penicillin, 0.1 µg streptomycin, and 0.25 µg mL−1 amphotericin), followed by two PBS washes.
Tissue was treated with dispase I (4 U mL−1; Sigma, Dorset, UK) overnight at 4 °C. The subsequent day, esophagus tissue was cut longitudinally, and scientists removed the epithelial layer.
The residual stromal tissue was cut and diced into small parts, and treated for 2 hours with sterile filtered collagenase type II dissolved in Hanks solution (100 U mL−1; Gibco, UK) in a 37 °C water bath.
Short-term vortexing occurred every 20 minutes. Tissue placed in collagenase II solution was doused in media containing Dulbecco’s modified Eagle’s medium (DMEM) high glucose supplemented with 10% FBS, 2 × 10−3 M LG, and A&A (100 U penicillin, 0.1 µg streptomycin, and 0.25 µg mL−1 amphotericin), and then filtered through a cell strainer with a 70 µm pore size.
The cell solution was centrifuged at 1400 rpm for 3 minutes, the supernatant removed, and the fibroblasts (1 × 106 cells) were plated out per T75 flask with the media that was previously used for dousing. Incubation of the cells occurred at 37 °C, and a change in media took place after reaching 48 hours and around every 2 days as required after that.
Oesophageal Cell Culture with Peptide Hydrogels: Peptide hydrogels were pipetted into 24-well ThinCerts from Griener, UK. Overnight, they were pre-equilibrated in media, to pH 7. Then, media were removed, and cells were ready to be introduced following specific protocols, as mentioned below.
Oesophageal Cell Culture with Peptide Hydrogels—2D Culture of OECs: OECs were trypsinized, and preparations were made for a cell suspension at a density of 3000 cells mm−2 /20 µL.
The cell suspension was seeded right on top of hydrogels at 100 µL volume, and the incubation period was for 30 minutes prior to submersion with media by adding to the surrounding areas of the insert in the well.
Cell–gel constructs were expanded until day 3, and media were changed every day. The controls used were gelatin-coated-glass coverslips.
Oesophageal Cell Culture with Peptide Hydrogels—3D Culture of OSFs: OSFs were trypsinized, and preparations were made for a cell suspension at a density of 200,000 cells/10 µL.
From the buffered hydrogels, media were removed, and the cell suspension was injected via a 10 µL pipette tip into the 150 µL of hydrogel to give a concluding seeding density of 200,000 cells/150 µL volume of the hydrogel.
Next, the cells were homogeneously integrated by combining the cell suspension together with the hydrogel via a pipette tip. OSF-laden hydrogels had an incubation period of 15 minutes to allow the hydrogel network to reassemble and then immersed in media.
Changes of media occurred every 3–4 days, and cell–gel constructs were cultured up to days 7 and 14. Controls used were collagen hydrogels (Rat tail, type I collagen, received from Millipore, UK).
Oesophageal Cell Culture with Peptide Hydrogels—3D Co-Culture: The peptide hydrogels with optimal performance for 2D culture (PGD-AlphaProC) and 3D culture (PGD-C2) were continued forward for the subsequent experiment.
Two systems were examined: system 1 (PGD-AlphaProC for both cell types) and system 2 (PGD-C2 combined with OSFs and PGD-AlphaProC with OECs on top). OSFs (500,000 cells/10 µL) were incorporated into PGD-AlphaProC or PGD-C2 peptide hydrogels that had been buffered, using the method previously explained.
After day 5 of culture, 50 µL of PGD-AlphaProC was added on top of PGD-AlphaProC (system 1) and PGD-C2 (system 2).
The incubator was set at 37 °C, and the stromal fibroblast media were added on top of the peptide hydrogels for 2 hours to be buffered. Media were removed, and OECs (3000 cells mm−2/20 µL) were seeded on top of the hydrogels, and the incubation period was for 2 hours to give time for attachment.
Stromal fibroblast media were removed from the top, and fresh stromal fibroblast media were added surrounding the inserts. This allowed the top layer to be cultured at the air-liquid interface. Composites were cultured at 37 °C until days 7 and 14, with changes in media occurring every 3–4 days.
OEC Viability Analysis on Peptide Hydrogels: Cell viability with hydrogels was examined utilizing a Live/Dead cell double staining kit, from Sigma in Dorset, UK. The removal of media from cell–hydrogel samples and controls occurred.
The media were washed two times with PBS and stained with calcein-AM and propidium iodide solution, as suggested by the manufacturer’s directions.
Samples were momentarily immersed in staining solution, and the incubation period was for 15-20 minutes at 37 °C. The staining solution was removed from samples, washed two times with PBS and imaged using a confocal microscope utilizing 490 nm excitation and 515 nm emission for calcein-AM and 535 nm excitation and 617 nm emission for propidium iodide.
Semi-quantification calculations of epithelial coverage on peptide surfaces were done using an ImageJ plug-in and Live/Dead staining images. The threshold was briefly adjusted to distinguish the difference between green and black, and then analysis was done to quantify percentage coverage.
OEC Metabolic and Proliferation Activity on Peptide Hydrogels: The alamarBlue assay (5 × 10−3 m, prepared in PBS; Sigma, Aldrich, UK) was used for the assessment of metabolic activity of OECs.
Media were removed from samples, washed two times in PBS, where they were immersed in PBS for 5 minutes each time, and then kept at an incubation period of 2 hours in alamarBlue solution at 37 °C.
Absorbance data were recorded after 2 hours in the culture at 570 nm excitation and 585 nm emission via a BMG LABTECH plate reader. alamarBlue solution was removed, and samples were systematically ashed in PBS and cultured in fresh media in preparation for the next time point.
Migration Assay: 12 well ThinCerts were covered with 200 µL of hydrogel (PGD-AlphaProC, PGD-CGD2, and collagen) and overnight, were buffered in media inside a 12-well plate.
A cloning ring and barrier created from a snipped pipette tip was used for cell seeding and positioned centrally on top of the hydrogel. OECs were trypsinized, and preparations of a cell seeding solution of 60,000 cells/50 µL were done.
The cell solution was seeded on top of the hydrogel surrounding the outside perimeter of the barrier. Constructs were cautiously incubated for 1 hour at 37 °C to give time for cell attachment to the hydrogel.
Media were added around the insert, and samples then incubated overnight. On a subsequent day, the barrier was removed, media were changed, and the samples were transferred for analysis to the JuLi Br & FL station microscope (NanoEnTek) in the incubator.
72-hour cell migration was monitored via time-lapse imaging. ImageJ was used to calculate cell migration rate and distance.
Characterization of Marker Expression of OECs Cultured on Peptide Hydrogels: Media were removed from OECs cultured for 3 days on top of hydrogels. Using PBS, hydrogels were washed and positioned in 10% formalin solution (neutral buffered; HT5012, Sigma-Aldrich, UK) at RT for 10–15 minutes.
Fixative was removed, and hydrogels were treated for 30 minutes at RT with permeabilization/blocking buffer (0.1% Triton X-100, 1% bovine serum albumin (BSA), and 1% goat serum, in PBS). Marker expression was evaluated via the procedures below.
Characterization of Marker Expression of OECs Cultured on Peptide Hydrogels: ZO-1: Treatment of hydrogels were done with 2.5 µg mL−1 primary ZO-1 antibody (rabbit ZO-1 anti-mouse; 61–7300, Invitrogen, UK). This was diluted in the same blocking buffer for 2 hours at RT.
Hydrogels were washed with PBS two times and treated with a goat anti-rabbit Alexa Fluor 546 secondary antibody (5 µg mL−1, catalog #: A11010; Invitrogen, UK) for 45 minutes at RT.
When the treatment was complete, hydrogels were washed with PBS two times and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 minutes at RT. Fluorescence microscopy (Nikon Eclipse 50i Microscope) was used to sample the images.
Characterization of Marker Expression of OECs Cultured on Peptide Hydrogels: Cytokeratin: Hydrogels were treated with mouse monoclonal anti-pan cytokeratins combined with fluorescein isothiocyanate (FITC) (1:50, catalog #: F3418, Sigma, UK) overnight at 4 °C.
On a subsequent day, samples were washed with PBS two times and counterstained with DAPI for 10 minutes at RT. Fluorescence microscopy (Nikon Eclipse 50i Microscope) was used for imaging.
Histological Staining of 3D Co-Cultures: Composites cultured for 7 and 14 days were positioned in 10% formalin for 1 hour and washed with PBS two times. Then, composites were processed overnight via a VIP 2000 (Vacuum Infiltration Processor, Miles Scientific).
Paraplast paraffin wax (Sigma) was used, and the sample was embedded into it to be divided transversely using a Leica RM2145 microtome. Samples were divided at 5 µm and attached to Poly-l-lysine slides (Thermo Scientific).
Histological Staining of 3D Co-Cultures: H&E Staining: Xylene was used for 5 minutes to de-wax the sections, followed by re-hydration via descending grades of ethanol to water. They were stained with hematoxylin for 5 minutes and “blued” in running water for 5 minutes.
Sections were stained in Eosin Y for 2 minutes and then washed three times for 1 minute in 95% ethanol. Sections were then washed in absolute ethanol three times and “cleared” in xylene. Next, they were mounted with DPX mountant (Sigma), and a LEICA DMRB fluorescence microscope was used to image samples.
Histological Staining of 3D Co-Cultures: Involucrin and Collagen Type I Immunohistochemical Staining: Xylene was used for 5 minutes to de-wax the sections, followed by thorough washing in descending grades of ethanol.
They were then positioned into methanol/H2O2 for 30 minutes at RT to block any endogenous peroxidase and finally rinsed in distilled water for 5 minutes.
Citrate pH 6 buffer was used for antigen retrieval with an incubation period of 30 minutes at 96 °C. Then, sections were blocked using R.T.U horse serum for 10 minutes at RT.
Primary antibody treatment (rabbit involucrin antimouse: 1:1000, overnight at 4 °C; catalog #: Biolegend-924401; Biolegend, USA) or (rabbit Collagen I anti-rat: 1:500, overnight at 4 °C; catalog #: ab34710; Abcam, UK) came before a secondary antibody treatment with R.T.U ImmPRESS anti-rabbit immunoglobulin G (IgG) (catalog #: Vector-MP-7401) for 30 minutes at RT.
Sections were treated with 3,3′-diaminobenzidine (DAB) substrate (Vector) for 5 minutes, and Nuclear Fast Red (Vector) was used for counterstaining for 2 minutes.
Sections were then de-hydrated to xylene and coverslips mounted with DPX mountant (Sigma). Fluorescence microscopy (Nikon Eclipse 50i Microscope) was used to image samples.
Histological Staining of 3D Co-Cultures—AE1/AE3 Immunohistochemistry Staining: Xylene was used for de-waxing for 5 minutes, before washing in descending grades of ethanol.
Sections were then positioned in methanol/H2O2 to block any endogenous peroxidase for 30 minutes at RT and then rinsed in distilled water for 5 minutes. Proteinase K was used for antigen retrieval at 37 °C for 10 minutes.
Then, sections were blocked in 2.5% goat serum for 10 minutes at RT, before being stained with primary antibody (mouse-IgG1 AE1/AE3 anti-rat, 1:20 dilution, overnight at 4 °C; catalog #: Thermo-MA1-82041; ThermoFisher, UK).
When the primary antibody had been removed, sections were treated with secondary antibody (1:200, biotinylated goat anti-mouse IgG) for 30 minutes at RT.
Sections were treated with avidin-biotin complex (Vector, UK) for 30 minutes at RT, rinsed in PBS for 5 minutes and treated with DAB substrate (Vector, UK) for 5 minutes.
Nuclear Fast Red (Vector, UK) was used for counterstaining of sections for 2 minutes, and samples were dehydrated to xylene and cover-slipped using DPX mountant (Sigma). Fluorescence microscopy (Nikon Eclipse 50i Microscope) was used to image samples.
Quantification of Collagen Type I Enzyme-Linked Immunosorbent Assay (ELISA): Media were removed from hydrogels (PGD-AlphaProC and PGD-C2), combined with rOSFs and cultured for 7 and 14 days.
Samples were rinsed with PBS and stored at −80 °C. Solubilization of collagen was completed using acetic acid by breaking up the cell–gel constructs (0.05 M acetic acid) and were transferred to a microcentrifuge tube, with an addition of 0.1 mg mL−1 pepsin solution.
Collagen digestion occurred for 24 hours at 4 °C with mild mixing on a rotor with sporadic forceful mixing by hand. Samples were then centrifuged (10,000 rpm for 3 minutes), and supernatants collected and transferred into collection tubes.
Supernatants were stored and analysis data collected to detect collagen type I, as suggested by manufacturers’ directions. This was done using ELISA (product code: 6013, Rat, Chondrex, USA). The BMG LABTECH plate reader was used to read optical densities (ODs) of ELISA samples at 490 nm.
Statistical Analysis: Calculations of mean and standard deviation (SD) were collected from three repetitions on each sample, apart from alamarBlue quantification data, where triplicate readings were done for each repeat.
Error bars on graphs show SD in positive and negative orientation. Data were tested for normality and a one-way, as seen in Figure 2C, or two-way, as seen in Table 1, Figures 2D, 4B, C. An analysis of variance (ANOVA)/ Kruskall–Wallis test was done, then a Tukey’s posthoc test to determine the significance of origin for all data. Significance levels were defined as: *p < 0.05, **p < 0.01, and ***p < 0.001.
References
[1] R. C. Fitzgerald, Gut 2006, 55, 1810.
[2] C. P. Wild, L. J. Hardie, Nat. Rev. Cancer 2003, 3, 676.
[3] R. Londono, S. Badylak, Tissue Eng., Part B 2015, 21, 393.
[4] J. Mannath, K. Ragunath, Ther. Adv. Gastroenterol. 2010, 4, 275.
[5] P. M. Shah, H. Gerdes, J. Gastrointest. Oncol. 2015, 6, 20.
[6] A. Nieponice, K. McGrath, I. Qureshi, E. J. Beckman, J. D. Luketich, T. W. Gilbert, S. F. Badylak, Gastrointest. Endosc. 2009, 69, 289.
[7] B. Qumseya, W. David, M. McCrum, Y. Dong, M. Raimondo, T. A. Woodward, M. B. Wallace, H. C. Wolfsen, Am. J. Gastroenterol. 2014, 109, 369.
[8] K. Uno, K. Iijima, T. Koike, T. Shimosegawa, World J. Gastroenterl. 2015, 21, 7120.
[9] P. D. Siersema, Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 142.
[10] S. Badylak, D. A. Vorp, A. R. Spievack, A. Simmons-Byrd, J. Hanke, D. O. Freytes, A. Thapa, T. W. Gilbert, A. Nieponice, J. Surg. Res. 2005, 128, 87.
[11] S. Badylak, S. Meurling, M. Chen, A. Spievack, A. Simmons-Byrd, J. Pediatr. Surg. 2000, 35, 1097.
[12] S. F. Badylak, T. Hoppo, A. Nieponice, T. W. Gilbert, J. M. Davison, B. A. Jobe, Tissue Eng., Part A 2011, 17, 1643.
[13] M. T. Wolf, K. A. Daly, E. P. Brennan-Pierce, S. A. Johnson, C. Carruthers, A. D’Amore, S. P. Nagarkar, S. S. Velankar, S. F. Badylak, Biomaterials 2012, 33, 7028.
[14] H. Mori, H. Kobara, K. Rafiq, N. Nishiyama, S. Fujhara, M. Ayagi, T. Yachida, K. Kato, T. Masaki, World J. Gastroenterol. 2013, 19, 5195.
[15] F. Yang, D. Ma, Q. Cai, Z. Li, J. Gastroenterol. Hepatol. 2013, 28, 1795.
[16] S. Collaud, T. Warloe, O. Jordan, R. Gurny, N. Lange, J. Controlled Release 2007, 123, 203.
[17] D. A. Johnson, R. Ganz, J. Aisenberg, L. B. Cohen, J. Deviere, R. Foley, G. B. Haber, J. H. Peters, G. A. Lehman, Am. J. Gastroenterol. 2003, 98, 1921.
[18] S. R. Caliari, J. A. Burdick, Nat. Methods 2016, 13, 405.
[19] S. G. Zhang, D. M. Marini, W. Hwang, S. Santoso, Curr. Opin. Chem. Biol. 2002, 6, 865.
[20] Y. Hong, R. L. Legge, S. Zhang, P. Chen, Biomacromolecules 2003, 4, 1433.
[21] A. Markey, V. L. Workman, I. A. Bruce, T. J. Woolford, B. Derby, A. F. Miller, S. H. Cartmell, A. Saiani, J. Pept. Sci. 2016, 23, 148.
[22] L. A. Castillo Diaz, A. Saiani, J. E. Gough, A. F. Miller, J. Tissue Eng. 2014, 5.
[23] G. B. Schneider, A. English, M. Abraham, R. Zaharias, C. Stanford, J. Keller, Biomaterials 2004, 25, 3023.
[24] S. Wan, S. Borland, S. M. Richardson, C. L. Merry, A. Saiani, J. E. Gough, Acta Biomater. 2016, 46, 29.
[25] A. Saxena, H. Ainoedhofer, M. E. Hollwarth, J. Pediatr. Surg. 2009, 44, 896.
[26] J. Kalabis, G. S. Wong, M. E. Vega, M. Natsuizaka, E. S. Robertson, M. Herlyn, H. Nakagawa, A. K. Rustgi, Nat. Protoc. 2012, 7, 235.
[27] Y. Zhu, M. F. Leong, W. F. Ong, M. B. Chan-Park, K. S. Chian, Biomaterials 2007, 28, 861.
[28] A. J. Engler, S. Sen, H. L. Sweeney, D. E. Discher, Cell 2006, 126, 677.
[29] A. M. Das, A. M. M. Eggermont, T. L. M. ten Hagen, Nat. Protoc. 2015, 10, 904.
[30] Y. M. Chen, R. Ogawa, A. Kakugo, Y. Osada, J. P. Gong, Soft Matter 2009, 5, 1804.
[31] K. Yasuda, N. Kitamura, J. P. Gong, K. Arakaki, H. J. Kwon, S. Onodera, Y. M. Chen, T. Kurokawa, F. Kanaya, Y. Ohmiya, Y. Osada, Macromol. Biosci. 2009, 9, 307.
[32] D. Liu, T. Wang, X. Liu, Z. Tong, Biopolymers 2014, 101, 58.
[33] M. Ahearne, Interface Focus 2014, 4, 1.
[34] S. R. Peyton, A. J. Putnam, J. Cell. Physiol. 2005, 204, 198.
[35] K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf, S. C. Rizzi, Biomaterials 2010, 31, 8454.
[36] C. M. Lo, H. B. Wang, M. Dembo, Y. L. Wang, Biophys. J. 2000, 79, 144.
[37] J. B. Frokjaer, S. D. Anderson, S. Lundbye-Christensen, P. Funch-Jensen, A. M. Drewes, H. Gregersen, Neurogastroenterol. Motil. 2006, 18, 104.
[38] N. H. Green, B. M. Corfe, J. P. Bury, S. MacNeil, J. Visualized Exp. 2015, 99.
[39] R. C. Orlando, Best Pract. Res., Clin. Gastroenterol. 2010, 24, 873.
[40] B. L. Beckstead, S. Pan, A. D. Bhrany, A. M. Bratt-Leal, B. D. Ratner, C. M. Giachelli, Biomaterials 2005, 6217, 26.
[41] W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA 1997, http://imagej.nih.gov/ij/.
Acknowledgments
Produced from materials originally authored by Deepak Kumar1, Julie E. Gough1, Marie O’Brien1, Alberto Saiani1,2, Victoria L. Workman2, Jane McLaren3, Felicity Rose3, Lisa White3 and Krish Ragunath4 from:
- School of Materials, University of Manchester
- Manchester Institute of Biotechnology, University of Manchester
- Centre of Biomolecular Sciences, School of Pharmacy, University of Nottingham
- NIHR Nottingham Digestive Diseases Biomedical Research Unit, Queens Medical Centre Campus, Nottingham University Hospitals NHS Trust
About Manchester BIOGEL

Over 15 years ago, Professors Aline Miller and Alberto Saiani at The University of Manchester synthesised a self-assembling oligo-peptide with interesting gelation properties. This work started with a small grant from the University.
Over subsequent years, the team meticulously crafted and studied self-assembling peptides to perfect their platform technology and produce a range of hydrogels ideal for 3D cell culture. In 2014, due to a demand for their materials, our company, Manchester BIOGEL was founded to enable these hydrogels to be readily available to researchers wishing to create new opportunities in the high-growth fields of 3D cell culture, 3D bioprinting and medical devices. Since opening our doors, we have supported scientists in the UK and beyond to create optimal environments for a wide variety of cell types.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.