Spinal muscular atrophy (SMA) is an incurable genetic condition in which severe proximal limb hypotonia is progressive and symmetrical. The cognition and intellect are always unaffected and may be high.
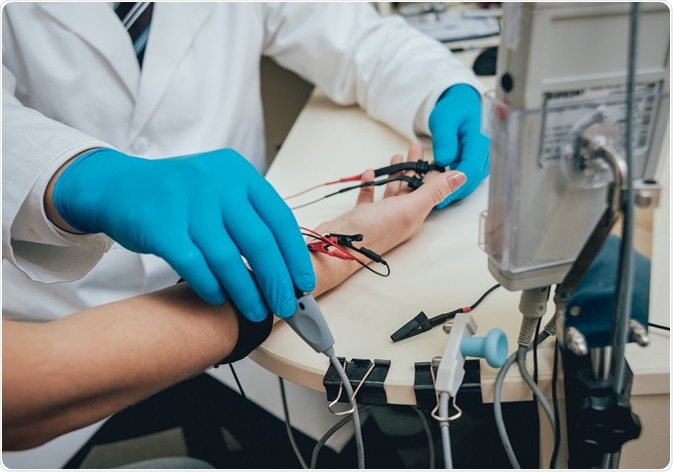
Medical examination using electromyography (EMG) Credit: Romaset/ Shutterstock.com
Clinical diagnosis
The clinical features are suggestive of the diagnosis. The severity of the condition is usually greater as the age of onset decreases, and hyporeflexia or areflexia is common in more severe forms of SMA. The characteristic features in the most severe form beginning in infancy include:
- Intact sensation
- Proximally predominant weakness, which progresses distally over time.
- Areflexia or hyporeflexia
- Weak cry
- Lack of head control
- Weak sucking
- Weak swallowing reflex
- Tongue fasciculations and atrophy
- Paradoxical breathing
One or more of these features may be more or less pronounced, depending on how complete the syndrome is, and this will also determine how early the age of onset is and the maximum motor function achieved.
Understanding Spinal Muscular Atrophy (SMA)
Diagnostic tests
The first test in such a patient is a genetic test to detect the SMN1 gene homozygous deletion, namely, a missing SMN1 exon 7 with or without a missing exon 8. This is almost 95% sensitive and 100% specific.
If this test is negative, but the clinical features strongly suggest SMA, other tests to confirm the presence of a motor neuron disease include:
- Creatine kinase assay
- Electromyography (EMG)
- Nerve conduction study
The EMG shows features such as:
- Active denervation
- Compensatory reinnervation
- Motor unit action potential (MUAP) enlargement
- Drop out and enlarged MUAPs on voluntary EMG
- Fibrillation potentials
- Short low-amplitude MUAPs
Nerve conduction studies show evidence of chronic loss of motor nerve axons and preserved sensory function. It is important to note that muscle biopsy is not performed for the diagnosis of SMA.
Once the presence of a motor neuron disease is confirmed on EMG, second-line genetic testing includes semiquantitative assays such as the real-time polymerase chain reaction (PCR) and Multiplex ligation-dependent probe amplification (MLPA). These detect the number of SMN1 gene copies. This increases the overall detection sensitivity to 98%.
If one copy of the gene is found on dosage analysis, the next step is to sequence the existing single allele to identify the mutation that is present and is causing the disease.
This is also recommended for patients born to consanguineous parents and genetically isolated pools and who show the typical clinical features but have two SMN1 alleles. Possibilities include having one allele mutated, having a double mutation of two alleles on the same chromosome, or even the appearance of a de novo mutation of the SMN1 gene.
If the diagnosis is confirmed, the copy number is also determined for the SMN2 gene, because this modifies the severity and prognosis. Most normal individuals have between 0 and 3 SMN2 gene copies. The copy number is not an infallible predictor of progression and prognosis, so care should be taken while counseling presymptomatic individuals.
If testing is negative, but the clinical diagnosis is SMA, it is quite possible that no mutation will be found in either the SMN1 or SMN2 exons, especially in type III SMA, which may be caused by deep intronic gene defects. However, other motor neuron diseases must be ruled out, such as X-linked SMA, distal SMA or juvenile amyotrophic lateral sclerosis.
If the EMG is negative for motor neuron disease, other conditions must be ruled out by full family history and repeating the complete examination and relevant tests. These may include:
- Congenital myopathies
- Congenital myotonic dystrophy
- Metabolic myopathies
- Myasthenic syndromes
- Congenital motor neuron and nerve diseases
- Other conditions such as Prader-Willi syndrome or neonatal sepsis
Useful tests would include CK assay, EMG, nerve conduction and MRI of the brain, metabolic parameters, with muscle biopsy and genetic testing when indicated.
Prenatal diagnosis and carrier testing
SMA is a prevalent diagnosis among genetic disorders, with its carrier frequency about 1 in 50. Thus, carrier testing would help in reproductive decision-making. It is based upon MLPA or real-time PCR, both semiquantitative assays, with the capability of diagnosing 93-95% of carriers with a single deletion of the SMN1 gene. It is usually performed only for a couple who are planning to conceive or are of reproductive age.
Couples undergoing such testing should go through the genetic counseling process to determine the risk of bearing a child with SMA. They should know that rare mutations, de novo mutations and the presence of both mutated copies of the SMN1 gene on the same chromosome, for instance, are not detected by this test but may cause SMA. In individuals at risk for carrier status because of having siblings or children with SMA, the at-risk individual is tested first, followed by the partner.
Prenatal diagnosis is performed only if both partners are carriers or have had an affected child previously. It is done by chorionic villus sampling but still cannot tell how severely a child may be affected, and thus the prognosis cannot be established. Thus, newborn screening is now being presented as a preferable option to identify affected children before becoming symptomatic and improving their quality of life.
In type I SMA, identifying the defect needs to be done very early as therapy should begin within weeks of birth. This is because more than 95% of motor neuron units are destroyed at 6 months of age.
Further Reading
Last Updated: Mar 1, 2021