Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has quickly begun to mutate since its outbreak in December 2019 in Wuhan, China. Mutations are common in coronaviruses, and SARS-CoV-2 has been found to have many different clades.
These clades can give scientists information on where certain strains of the virus are concentrated, and how these different clades may impact the virulence of SARS-CoV-2, the speed of the disease spread, and its resistance to antiviral medications.
 Image Credit: Andrii Vodolazhskyi/Shutterstock.com
Image Credit: Andrii Vodolazhskyi/Shutterstock.com
These clades can give scientists information on where certain strains of the virus are concentrated, and how these different clades may impact the virulence of SARS-CoV-2, the speed of the disease spread, and its resistance to antiviral medications.
What is a clade?
A clade is a term for a group of organisms that all originate from a common ancestor, and is widely used in biology. Using phylogeny, which is the evolutionary history of a group of organisms, the development of changes in a set of descendant organisms can be tracked.
In virology, a clade describes groups of similar viruses based on their genetic sequences, and changes in those viruses can also be tracked using phylogeny. Rapid genome sequencing is the method by which developments in a virus’s genomic makeup can be tracked.
SARS-CoV-2 is itself a clade within the family coronaviridae and the genus betacoronavirus. Generally, the genetic variations of a virus are grouped into clades, which can also be called subtypes, genotypes, or groups.
Rapid genome sequencing can help to quickly work out where a person has become infected with a certain clade of SARS-CoV-2. For instance, the first four cases of COVID-19 in New South Wales, Australia, were found to be closely related to the dominant strain of SARS-CoV-2 found in Wuhan, and these first four cases were all in people who had recently returned from traveling in China. This meant that travel could then be restricted between China and Australia to limit the numbers of infected people traveling to and from the two countries.
As discussed in a Virus Evolution paper, published in April 2020, cases were also tracked from Australia to Iran, where it was found that the genomes were all from one monophyletic group characterized by three nucleotide substitutions in the SARS-CoV-2 genome, when compared to the prototype strain from Wuhan.
Evolution of the SARS-CoV-2 genome
An investigation published in June 2020 and carried out by the World Health Organization (WHO) showed how the SARS-CoV-2 genome has evolved as it has spread across the world. This investigation did not show how these evolutions changed the virulence of the virus, but it did show that the most common SARS-CoV-2 clade was the D614G variant within the six clades and 14 subclades it identified.
The investigation included 10,022 SARS-CoV-2 genomes from 68 different countries. In total, WHO detected 65,776 variants and 5,775 distinct variants, which comprised:
- 2,969 missense mutations
- 1,965 synonymous mutations
- 484 mutations in non-coding regions
- 142 non-coding deletions
- 100 in-frame deletions
- 66 non-coding insertions
- 36 stop-gained variants
- 11 frameshift deletions
- Two in-frame insertions.
Since then, numbers will have increased.
The D614G variant is located in the B-cell epitope and has been found to have a very immunodominant region, which may affect how well a vaccine may work against it.
The largest clade found in the WHO study was D614G, which had five subclades associated with it. The non-coding variant 241C > T, along with 3037C > T, and ORF1ab P4715L were found in most of the samples in the D614G clade.
Additionally, almost every strain that had a D614G mutation featured mutations in proteins that control viral replication, which has implications for how quickly the virus can multiply. This particular protein is what anti-viral drugs remdesivir and favipiravir target. It could be possible that strains of SARS-CoV-2 quickly become resistant to drugs that target these proteins.
The second-largest clade identified by the WHO study was L84S, which comprises two subclades. L84S was a clade found in people traveling from Wuhan in the early phases of the SARS-CoV-2 outbreak.
What are the different SARS-CoV-2 clades?
There are multiple different nomenclatures for the SARS-CoV-2 clades. Each health organization may use its own identifier for different variants.
PANGOLIN nomenclature
The Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software team proposed nomenclature for the SARS-CoV-2 clades in a Rambaut et al. (2020) article. This contains the main lineages of A, B, B.1, B.1.1, B.1.177 and B.1.1.7. These lineages divide up further.
The generally used names of strains e.g. the B.1.1.7 (British originating) and B.1.351 (South African originating) strain derive from this.
A is the original strain used as a reference sequence.
The GISAID nomenclature
There are many thousands of complete and high-coverage genomes available on the Global Initiative on Sharing Avian Influenza Data (GISAID).
GISAID separates the clades of SARS-CoV-2 into S, O, L, V, G, GH, GR, GV, and GRY.
The S and L clades were around at the beginning of the pandemic. S continued to be prevalent initially whilst L split into G and V. G further split into GR and GH, and then later GV. GR split into GRY after July 2020. The letters come from the mutations that caused them to branch.
The G clade is equivalent to PANGO B.1 lineage, with GR representing PANGO B.1.1 lineage. The S clade is equivalent to PANGO A lineage (the original virus, sequence zero, used as the reference), and the V strand is PANGO B.2 lineage. L is another eaerly lineage. O stands for others that do not match the genetic criteria of the other clades.
The G clade and its subsequent branches include the S-D614G strain.
Current GISAID data on the phylogenetic tree and geographical and temporal distribution of these clades can be viewed here.
At the beginning of the pandemic, in January 2020, the L, S and O clades had the majority. These all decreased as the G clade and its descendants increased in proportion over the next year. As of March 2021, the L, S and O clades constitute almost nothing, with the new GRY clade taking up the biggest proportion of the G clades.
The GRY clade represents the B.1.1.7 strain that originated in Britain and has since spread across the globe to over 90 countries.
Nextstrain nomenclature
Alternatively, Nextstrain divides the SARS-CoV-2 strains into 19A, 19B, 20A, 20B, 20C, 20D, 20E, 20F, 20G, 20H, 20I, 20 J. Phylogenetic trees and geographical/ temporal maps can be viewed for these classifications here.
Within these clades, 19B is the original reference strain. 20I/501Y.V1 refers to the B.1.1.7 variant that originated in Britain; 20H/501Y.V2 refers to the B.1.351 strain that originated in South Africa; and 20J/501Y.V3 refers to the P.1 strain that originated and spread from Brazil.
Variants of concern
There are currently selected ‘variants of concern’ which have originated in some geographical locations and since spread globally due to mutations causing increased transmissibility. They are causing concern due to wide spread and reduced efficacy of some vaccines or antibody responses across some of the strains.
These include the B.1.1.7 strain, the B.1.351 strain and the P.1 strain (using PANGOLIN lineage terminology).
The WHO recently announceda new naming system for these variants in order to provide a more practical and easier way of referring to them, particularly in non-scientific contexts. This naming system uses Greek lettering.
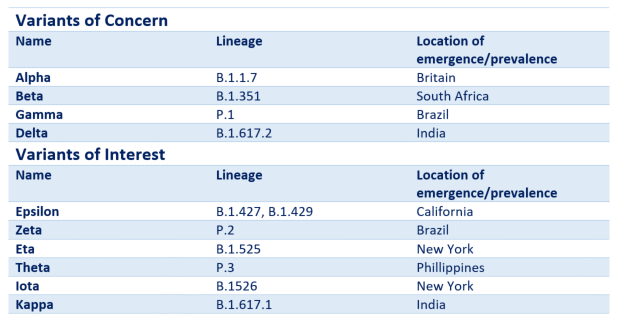
Table 1. The Greek lettering system for naming SARS-CoV-2 variants and their PANGOLIN lineage equivalents.
Summary
SARS-CoV-2 has proven to be a genetically diverse virus that has now become endemic within humans. Identifying and tracking clades that become dominant in certain geographical regions may inform the development of effective vaccination, as certain anti-viral drugs or vaccines may not work or be as effective against mutated forms of the virus, or clades of the virus that have developed resistance through genomic mutation.
There are multiple different naming systems for the clades of SARS-CoV-2, but all of them are rooted in the same genetic analysis and allow tracking of different variants geographically and understanding of how they have evolved.
References
Further Reading
Last Updated: Mar 18, 2026
 COVID-19 initially reduced asthma attacks but widened socioeconomic disparities, study finds
COVID-19 initially reduced asthma attacks but widened socioeconomic disparities, study finds