Aniridia is a rare, vision-threatening condition defined by partial or complete hypoplasia of the iris. It is a serious genetic disorder associated with the abnormal development of the iris of the eye. It often affects both the eyes and is usually present since birth. It can also affect various other parts of the eye including the cornea, retina, lens, anterior chamber, and optic nerve.
Several types of aniridia have been identified over the years. Each type is recognized according to the symptoms present in the patient. Aniridia can be either present since birth or acquired later in life. It may be linked with multiple ocular morbidities and several systemic diseases.
 Aniridia - a rare genetic eye disorder that affects the iris. Image Credit: Sruilk / Shutterstock
Aniridia - a rare genetic eye disorder that affects the iris. Image Credit: Sruilk / Shutterstock
Causes and symptoms
No exact cause of aniridia has been identified so far. However, according to research studies, most forms of aniridia are linked with the mutation of the PAX6 gene. PAX6 is associated with the health and development of the eye. Mutation in PAX6 leads to the incorrect development of the eye causing aniridia.
The disorder usually exhibits an autosomal dominant pattern of inheritance, with some cases being sporadic. Aniridia can occur as an isolated eye complication. It can also be present as a part of the WAGR (Wilms tumor-aniridia-genital anomalies-retardation) syndrome.
Aniridia is characterized by the (full or partial) absence of the iris of the eye. Some patients with mild forms of aniridia have their vision intact. This disorder develops when the iris of one or both eyes fail to form correctly before birth. Aniridia is usually visible from birth.
Aniridia can be a solitary aberration or one of many symptoms associated with a more serious illness. Other complications like keratopathy and glaucoma may also be present leading to visual impairment in patients.
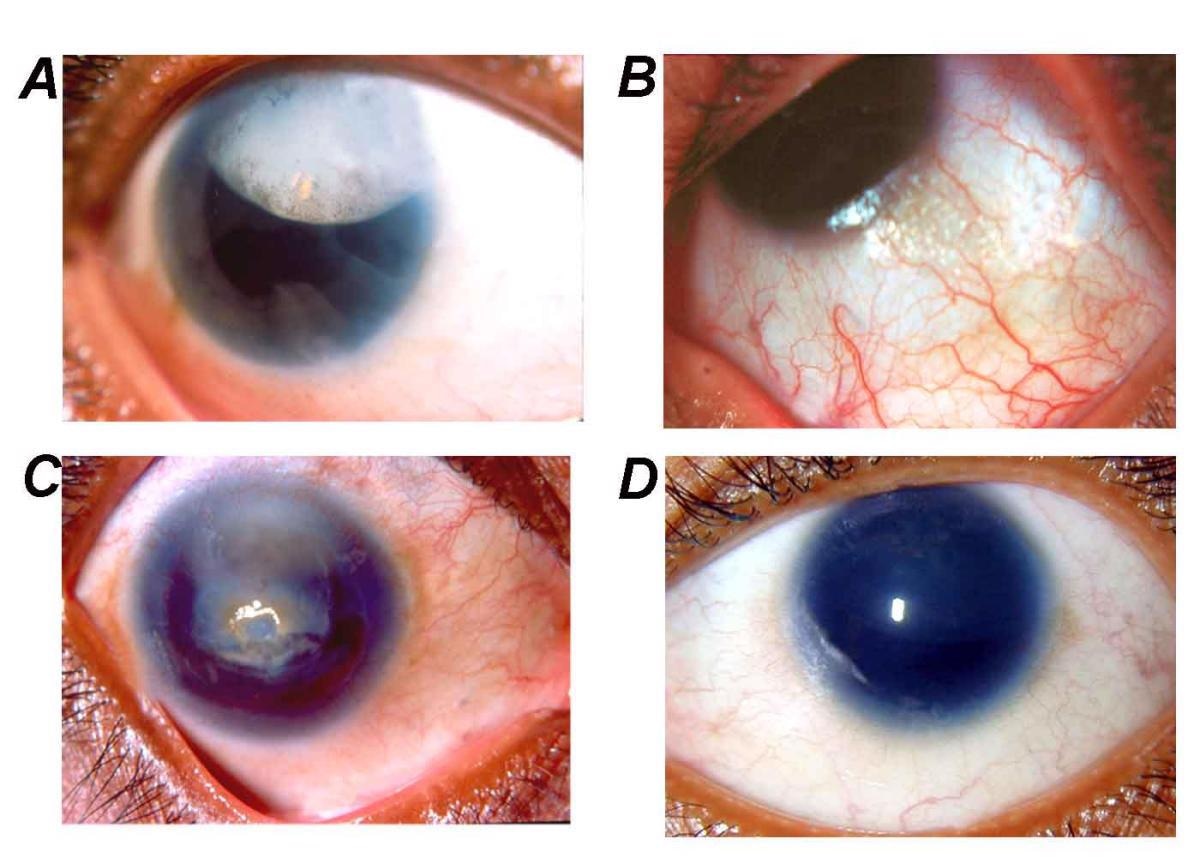
By Neethirajan G, Krishnadas SR, Vijayalakshmi P, Shashikant S, Sundaresan P. PAX6 gene variations associated with aniridia in south India. BMC Med Genet. 5, 9. 2004. PMID 15086958. DOI:10.1186/1471-2350-5-9, CC BY 2.0, https://commons.wikimedia.org/w/index.php?curid=3121372
Forms of aniridia
Classic/isolated
Isolated aniridia is the partial or complete lack of the iris. Some patients with this form of aniridia may not experience any complications of the eye as the pupil seems to be normal and usually, only a single eye is mildly affected. Severe forms of isolated aniridia cause vision impairment later in life.
Associated symptoms usually involve cataracts, corneal pannus, and glaucoma. In some cases, nystagmus and foveal hypoplasia may also occur causing blurry/cloudy vision.
WAGR Syndrome
Robert Miller et al. published the results of a review of the case records of 440 people with Wilms tumor, a rare embryonic cancer that affects the kidney, in 1964. Six of these children (1.4 percent) also had aniridia. This seemed highly unlikely to be a coincidence given the 1 in 50,000 live births incidence of aniridia at the time. Many more cases were published after this original report.
Depending on whether Wilms tumor is present or not, this constellation of clinical symptoms is referred to as WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, mental retardation) or AGR syndrome.
Gillespie syndrome
The characteristic features of Gillespie syndrome include partial aniridia, non-progressive cerebellar hypoplasia, ataxia, congenital hypotonia, and intellectual disability. Due to aplasia of the tissue central to the collarette, the iris defect in GS is bilateral and has a distinct scalloped look.
The aberrant development of the ectoderm-derived sphincter musculature and the surrounding mesoderm-derived stromal tissues at the distal tip of the fetal iris appears to be the cause of the aplasia found in GS. Unlike PAX6-associated aniridia, the ocular abnormalities in GS are primarily limited to the iris and the presence of iridolenticular strands. Foveal hypoplasia is a rather uncommon occurrence.
Genetics
In around 90% of aniridia cases, the function of one copy of the PAX6 gene is lost. In this, intragenic mutations account for two-thirds, and chromosomal rearrangements account for one-third. Approximately two-thirds of all instances are familial, with dominant inheritance and significant penetrance.
For sporadic cases, the novel mutation (which presumably occurred de novo in one parent’s germline) will be dominantly inherited in the following generations. PAX6 is found in the developing eye, different parts of the brain, the neural tube, the gut, and the pancreas.
It plays many roles in the development of the retina, lens, cornea, and iris, and is active early in ocular morphogenesis. Mutations of other genes including FOXC1, PITX2, and PITX3 can also lead to aniridia-like phenotypes.
Epidemiology
Aniridia is found in around 1.8 out of every 100,000 newborns. Reports show an equal number of cases for both genders.
Non-ocular features of the disease
Recent research shows that patients with aniridia may be suffering from non-ocular features including sensory and neurological complications. Reduced olfaction appears to be the most common functional disability in people with isolated aniridia.
MRI scans have reported abnormalities of the anterior commissure, cerebellum, corpus callosum, pineal gland, anterior cingulate cortex, temporal lobes, occipital lobes, and olfactory bulb. Cognitive function, on the other hand, is, however, normal. Behavioral issues and developmental delays are also uncommon.
Due to irregular interhemispheric transmission, aniridia patients may have central auditory processing impairments, which can cause hearing problems.
Diagnosis and treatment
The diagnosis process involves two approaches: clinical and genetic. Clinical diagnosis comprises of evaluation of medical history and examination by an ophthalmologist. Genetic diagnosis involves the assessment of familial history and genetic testing for the PAX6 gene. Genetic counseling can be beneficial.
The purpose of aniridia treatment is usually to improve and preserve vision. Glaucoma and cataracts may benefit from medications or surgery. In some circumstances, contact lenses may be advantageous. Patients should be assessed for the likelihood of developing Wilms' tumor if a hereditary etiology cannot be found.
The first artificial iris, a surgically implanted device to treat adults and children with aniridia, was authorized by the FDA in 2018. This device may aid in the reduction of light sensitivity and glare. It can also enhance the cosmetic look of the eye.
Aniridia is a challenging complication of the eye and requires life-long ophthalmologic care. More research and clinical studies are essential to facilitate a better understanding of the disease and to develop better therapeutic approaches.
References:
- Tripathy K, Salini B. Aniridia. [Updated 2021 Aug 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538133/
- (2020). Aniridia. [Online] National Organization for Rare Disorders. Available at: https://rarediseases.org/rare-diseases/aniridia/
- Hall, H. N., Williamson, K. A., & FitzPatrick, D. R. (2019). The genetic architecture of aniridia and Gillespie syndrome. Human genetics, 138(8-9), 881–898. https://doi.org/10.1007/s00439-018-1934-8
- (2016). Aniridia. [Online] NIH-National Center for Advancing Translational Sciences. Available at: https://rarediseases.info.nih.gov/diseases/5816/aniridia
- Hingorani, M., Hanson, I., & van Heyningen, V. (2012). Aniridia. European journal of human genetics: EJHG, 20(10), 1011–1017. https://doi.org/10.1038/ejhg.2012.100
Further Reading
Last Updated: Oct 9, 2023