Introduction
One of the major tasks in present-day biology is to map the chromosomal positions of nucleosomes, TFs, chromatin remodeling enzymes, histone modifications, polymerases and chaperones. Genome-wide profiling of histone alterations and DNA-binding proteins is performed using a method called chromatin immunoprecipitation followed by sequencing or ChIP-seq (Figure 1).
Chromatrap® offers a user-friendly and straightforward ChIP format in combination with next-generation sequencing (NGS) technology. It exhibits compatibility with low and high cell numbers, and has been tested to identify epigenetic marks and transcription factors in primary and secondary cell lines.
As Chromatrap® ChIP-seq is available in a 96-well plate, high-throughput analyses can be easily performed and coordinated, complicated epigenetic mechanisms and gene regulations can be evaluated on a global level. This article shows how the Chromatrap® ChIP-seq kit has been deployed for explaining the genome-wide binding arrangements of nuclear receptor binding occurrences in human adenocarcinoma cells, signifying the rapid multiplex capability in conjunction with massively parallel NGS. For genome-wide protein-DNA analysis, Chromatrap® ChIP-seq assays allow reliable, high-resolution, and low-noise amplification.
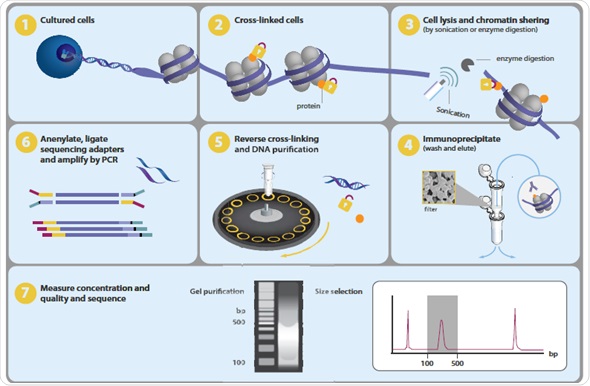
Figure 1.Overview of ChIP-seq process
Monitoring of the estrogen receptor (ER) functioning was performed using a cell line model to prove how the Chromatrap® ChIP-seq is capable of sensitively and selectively enhancing DNA for NGS.
This article focuses on ligand-independent and -dependent binding of ER in cells, extracted from human endometrial epithelial adenocarcinoma tumors. Further, with regards to ER binding, the relationship of epigenetic mark related to open chromatin (H3K4me3) and epigenetic mark related to closed chromatin (H3K27me3) is studied. Featuring the capability of carrying out analysis of several samples or targets, Chromatrap® ChIP-seq, in its high-throughput configuration, helps determining difficult and concurrent epigenetic profiles and TF complexes. In this particular configuration, Chromatrap enables multi-target, genome-wide screening across a range of cell types.
Methodology
Culture of 1x107 human endometrial epithelial adenocarcinoma cells was carried out for 48h in phenol red-free medium with 10% charcoal-removed FBS. The next step was a 4-hour-treament using 10nM 17β-estradiol (E2). Formulation of chromatin was carried out based on the Chromatrap® ChIP-seq spin column protocol. The immunoprecipitation needs to be validated at a particular genomic locus before sequencing. To achieve this, positive primers and antibody controls are offered by Chromatrap®. By means of qPCR, the immunoprecipitation enrichment was validated for ER and H3K4me3 antibodies for the XBP1 and GAPDH locus (Figure 2).
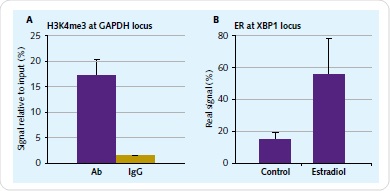
Figure 2. Chromatin IP validation. The enrichment of H3K4me3 associated with GAPDH (A) and ER associated with XBP1 (B) was validated prior to library preparation with a small-scale 2µg IP reaction.
In order to conduct sequencing with 30µg of chromatin and 5µg of antibody (ER, H3K4me3 or H3K27me3), immunoprecipitations were carried out. At the time of data analysis, an equal amount of IgG was used in another IP and deployed as a background control. Reverse crosslinking and purification of the chromatin was performed subsequent to immunoprecipitation by using the illustra GFX PCR DNA and Gel Band Purification Kit, followed by elution of the resulting chromatin in 50µl Tris-HCl elution buffer. The Agilent Bioanalyzer was used to validate ChIP DNA for corroborating the concentration and distribution of the sample size (Figure3).
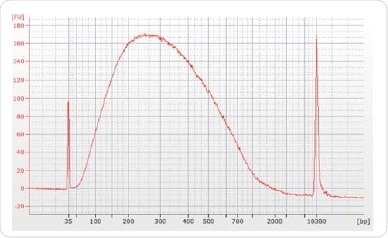
Figure 3. Size selection and library preparation. The majority of post ChIP sheared chromatin was approximately 200-300 bp. Chromatrap® ChIP-seq.
Quantification of the samples was performed with the Qubit 2.0 fluorometer, and based on the Illumina® TruSeq ChIP Sample Prep Kit protocol (IP-102-1012), library preparation was carried out. An initial starting quantity of 5-10ng ChIP DNA is recommended by Illumina®. For higher library complexity and lower probability of PCR replications, 25ng of DNA was used in this experiment.
The Chromatrap® ChIP-seq kit is configured such that it is compatible with Illumina® ChIP-seq library preparation kits and sequencing platforms to provide superior quality DNA for sequencing. Chromatrap’s ChIP-seq 96 option, distinct solid-phase matrix increases reproducibility and sample throughput, thanks to a matrix that eliminates blocking, low sample volumes, and centrifugal wash steps.
Chromatrap® ChIP-seq Data Analysis
The genomic positions of modified chaperones, histones, nucleosomes, DNA-binding enzymes and TFs are determined by computational mapping of the sequenced DNA to gain insights into the contribution of these protein-DNA interactions in gene expression as well as in other cellular processes (Mardis et al, 2007). Figures 4A, B and C show the FastQC values for the sequencing run and the further duplication levels acquired for ERα.
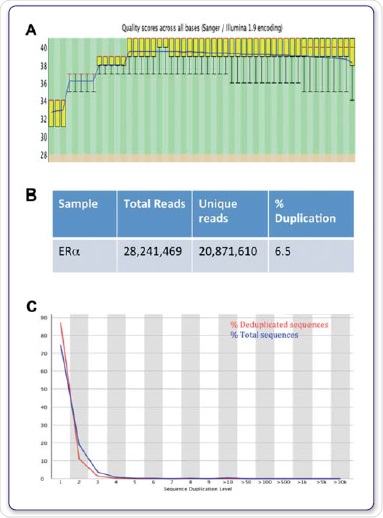
Figure 4. Sequence quality, total reads and duplication. FastQC was used to evaluate quality scores across all bases (A), total and unique reads (B), and the sequence duplication level (C).
In order to identify areas of enhancement, related IgG datasets were deployed as background controls. By means of the Illumina® HiSeq2000 with 1 x 50 bp reads, sequencing at high-throughput was carried out. FastQC was used to study the sequencing data quality. On an average, a Q30 score, correlating to a 0.001% probability of a base being labelled inaccurately was applicable to more than 97% of all data sets, as shown in Figure 4A.
Mapping of sequences to the human reference genome (USCS human genome assembly hg19) was performed and their alignment was carried out by means of Bowtie 2. Chromatrap® ChIP-seq Data Analysis software was applied to analyze the data using SICER or MACs for ER or H3K27me3 and H3K4me3, respectively (Figure 5).
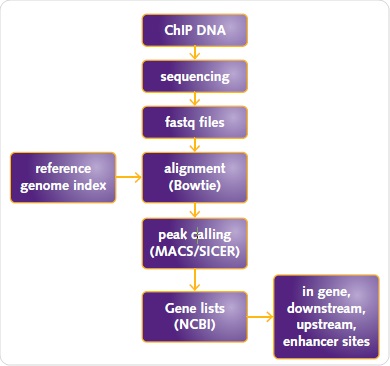
Figure 5. ChIP-seq data analysis workflow. ChIP DNA sequencing fastq files alignment (Bowtie) reference genome index peak calling (MACS/SICER) Gene lists (NCBI) in gene, downstream, upstream, enhancer sites.
Single end sequencing was implemented in this experiment and optimization of Chromatrap® was also performed with paired end sequencing, minimizing alignment uncertainty to the reference genome, especially in repetitive areas.
FastQC Sequencing Quality Parameters
There is a need for adequate sequencing depth for effective ChIP-seq data analysis. The needed depth is based on the genome size, and the size and number of the protein binding sites. Roughly, 20 million reads are needed for mammalian TFs that have thousands of binding sites. A very high number of TFs and histone marks have several binding sites and need 60 million reads for adequate coverage in mammalian ChIP-seq (Chen et al, 2012).
Results
Putative consensus sequences and nuclear receptor binding arrangements were revealed after obtaining adequate sequencing depth. In this experiment, using the unique reads (74%) acquired from Chromatrap® ChIP-seq, scientists were able to determine areas in the genome where their investigated protein is attached and can be used to expose putative consensus sequences (Figure 6).
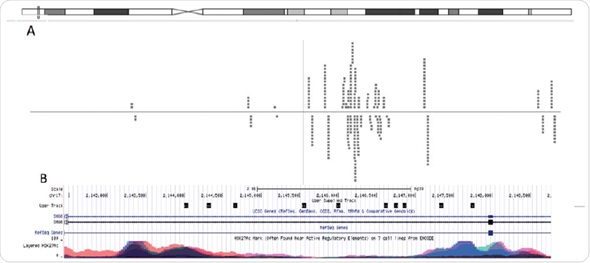
Figure 6. Graphical visualization of read alignment. When aligned to the genome using Bowtie software, MACSidentified peaks can be visualized through genome-browsers. (A) Chipster genome browser view of the chromosomal locus near the SMG6 gene. The dashed lines represents reads, which align to the reference genome that are associated with H3K4me3. (B) UCSC genome browser view of the same chromosomal locus overlayed with H3K27Ac ENCODE data.
Selectively Enriched Nuclear Receptor Binding
A complicated range of activation mechanisms are exhibited by ERs, which are typical targets for genome wide binding assays. For ER, binding mechanisms that are dependent and independent of ligands are clearly seen in the obtained genome-wide patterns. It was observed in the controlled untreated samples that ER was observed at 2377 genes, and there was an enhancement of this enrichment after ligand activation (Figure 7).
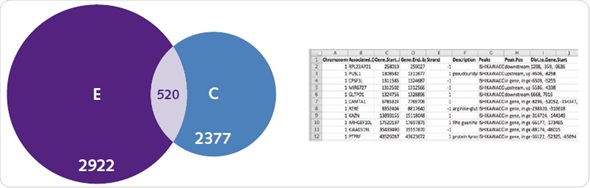
Figure 7. Ligand-dependent and -independent ERα binding. Chromatrap® ChIP-seq resolved uniquely identified genes associated with ER under both estradiol (left), and untreated control conditions (right), as well as genes commonly associated with both (center). In addition, subsequent gene ontology lists were generated that describe unique or commonly associated genes between control and E2 treatment.
Compilation of genes belonging to specific treatment groups results in a database, which consists of data about genomic loci, function of the gene, the beginning and ending position of the gene and whether the peak is observed downstream, upstream or inside the gene itself (Figure 7). Most of the genes related to the E2 group are associated with metabolic pathways and cell proliferation.
Nuclear Receptor Function and the Epigenetic Landscape
H3K4me3 is related to the open chromatin structure of active genes, most of which are constitutively active because of their role in important cellular processes. An assay of concurrent enrichment of ER and histone methylation marks related to open and close chromatin structures was performed to demonstrate the applicability of high throughput Chromatrap® ChIP-seq on multiple marks.
Under basal conditions, the number of genes typically related between ER and H3K4me3 or H3K27me3 were identified. It was observed that out of the 376 genes related to ER, around one-third are also related to H3K4me3 (Figure 8a). It is surprising to note that more genes were related to H3K27me3, which is an epigenetic alteration corresponding to silenced regions (Figure 8b). When compared to the peaks detected for ER, the peaks related to histone alterations were significantly higher.
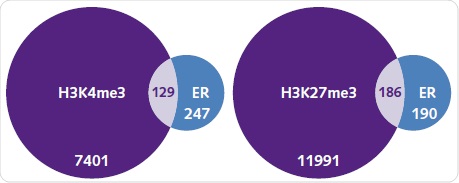
Figure 8. ER recruitment and the histone methylation landscape. The number of genes associated with H3K4me3 (left) and H3K27me3 (right) when compared with ER binding under basal conditions. Each diagram shows the number of unique and commonly shared genes between each group.
Conclusions
For the transformation of genome wide ChIP-seq assays into high throughput experiments that can map multiple marks concurrently, there is a requirement for innovative technologies and products. Complicated TF and epigenetic mechanisms that monitor gene regulation can be clearly understood using Chromatrap® ChIP-seq. This knowledge is useful for developing therapeutic strategies critical in both normal disease and physiology.
Also, standard sample preparation, evaluation and IP procedures are required for cohort and meta-studies so that large data sets can be archived in a normalized data format for common use and performing metadata analysis.
This article proves the capability of the Chromatrap® ChIP-seq to study genome-wide nuclear receptor binding in a complicated epigenetic landscape in cell lines that show multiple isoforms. Gaining insight into the dynamic nature of TF binding helps explaining the complex transcriptional gene regulation involved in the advancement of cancer.
More than 2000 accurate genomic locations of direct ER binding could be determined and also data on the relationship of histone alterations associated with silenced or linked chromatin areas were detected. The Chromatrap® ChIP-seq kit has chromatin loadings of up to 30µg, enabling a dynamic sample loading range. This allows lower amounts of chromatin to be loaded, thereby reducing the need for antibodies. This kit shows excellent compatibility with key sequencing platforms, such as the Illumina® HiSeq and MiSeq instruments.
About Chromatrap®
 Chromatrap® is a product of Porvair Sciences, a wholly owned subsidiary of Porvair plc. We are one of the largest manufacturers of Ultra-Clean microplates, 96 well well filtration plates and Microplate handling equipment for life science and synthetic chemistry. With offices and Class VIII clean room manufacturing located in the UK, combined with a world-wide network of distributors and dedicated distribution hub in the USA, we pride ourselves on our continuous innovation, research and flexibility to meet customer demands. We offer OEM production and contract manufacturing through our North Wales facility.
Chromatrap® is a product of Porvair Sciences, a wholly owned subsidiary of Porvair plc. We are one of the largest manufacturers of Ultra-Clean microplates, 96 well well filtration plates and Microplate handling equipment for life science and synthetic chemistry. With offices and Class VIII clean room manufacturing located in the UK, combined with a world-wide network of distributors and dedicated distribution hub in the USA, we pride ourselves on our continuous innovation, research and flexibility to meet customer demands. We offer OEM production and contract manufacturing through our North Wales facility.
Our porous polymeric material, BioVyon™, whose chemical functionalisation can endow it with internal surface properties individually configured to capture and separate target species out of difficult mixtures, has opened up many possibilities in the field of BioSciences where molecules of interest such as DNA, RNA, proteins etc can be selectively pulled out of complex mixtures of biological origin. The materials have proven to be a remarkably good substrate for accepting novel chemistries such as the organically bound Protein A and Protein G in Chromatrap®.
Using our 25 years experience of microplate manufacturing, Porvair Sciences has now developed a high-throughput bead-free ChIP assay based on our filtration plates containing our Chromatrap chemistry. Chromatrap-96 enables large scale epigenetic screening to become a reality in many laboratories and eliminates many of the long and laborious steps previously undertaken in such work.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.