As the global population ages, Alzheimer’s Disease (AD) is a neurodegenerative disease which is becoming more common. A cure has not yet been found, but over a century of research has enabled scientists to better understand what contributes to its development and how the disease progresses.
Brains with AD have two main features: neurofibrillary tangles consisting of a protein called tau and plaques consisting of a peptide called amyloid-beta. Tau aggregation and amyloid-beta and are both thought to contribute to AD, but there are different hypotheses regarding which aggregates first, this should be the target for treatments, and how the two proteins interact.
A Brief History of Alzheimer’s Disease Research
When Alois Alzheimer recorded “presenile dementia” in Auguste Deter, his 51-year-old dementia patient, he observed delusions and short-term memory loss which were common in patients in their 70s, but not patients in their 50s. He observed anomalies in her brain after her death: plaques and tangles.1
Even though plaques had been observed previously in other cases of dementia,2 Alzheimer was the first to note the tangles. Emil Kraepelin, his senior colleague, suggested he had found a new disease and called it Alzheimer’s Disease.
In the 1930s and 40s, instances of autosomal dominant, or early-onset familial AD, which are inherited from a parent with an abnormal gene and happen in patients younger than 65, were studied and documented.
This was thought to be a different disease than “senile dementia,” which had similar symptoms but did not follow an inherited autosomal dominant inheritance pattern and occurred in patients over the age of 65.3
![Über eine eigenartige Erkrankung der Hirnrinde,”which translates to “About a Peculiar Disease of the Cerebral Cortex.” The original case study by Alois Alzheimer was published in 1907. By © Foto H.-P.Haack [CC BY 3.0 (https://creativecommons.org/licenses/by/3.0)], via Wikimedia Commons.](https://www.news-medical.net/image-handler/picture/2020/1/Art-2-img-2-1.jpg)
“Über eine eigenartige Erkrankung der Hirnrinde,”which translates to “About a Peculiar Disease of the Cerebral Cortex.” The original case study by Alois Alzheimer was published in 1907. Image Credit: Foto H.-P.Haack [CC BY 3.0 (https://creativecommons.org/licenses/by/3.0)], via Wikimedia Commons
Researchers Blessed, Tomlinson, and Roth recognized that the pathology of early-onset AD was the same as that of “senile dementia” in the 1960s and 70s.4 AD was acknowledged as a widespread public health issue in aging populations, instead of a rare genetic disorder, so research funding and interest dramatically increased.
The Amyloid Hypothesis
Later in life, individuals with Down syndrome can often experience memory loss and neurological changes. This includes the formation of plaques similar to those observed in AD patients.5 Glenner and Wong isolated a 42 kDa peptide known as amyloid-β (Aβ) from the plaques of both Down syndrome and AD brains in 1984.6
They suggested that because Down syndrome and AD plaques both contain Aβ, the conditions share a common pathogenic process. In addition, they claimed that since Aβ is encoded in chromosome 21, and Down syndrome is a result of the trisomy of chromosome 21, the genes on chromosome 21 are responsible for AD pathology.6
In turn, this led to the amyloid hypothesis, which is also known as the amyloid cascade hypothesis or amyloid-beta hypothesis. It states that Aβ aggregation triggers the chain of events that ultimately result in AD pathology and symptoms.7
This chain of events includes the aggregation of hyperphosphorylated tau proteins into neurofibrillary tangles (NFTs), in addition to oxidative stress and inflammation which can result in neuronal dysfunction and death.
There were multiple conflicting and complex theories regarding the pathogenesis of AD at this time.7 The Down syndrome link was compelling, and the amyloid hypothesis was relatively simple and gave researchers a therapeutic target.7 This made it the focus of extensive research over the next few decades.
Evidence for the Amyloid Hypothesis
Alzheimer’s Genes and the Amyloid Hypothesis
The APP gene encodes the Amyloid Precursor Protein (APP) and is located on chromosome 21. When APP is cleaved by gamma-secretase the amyloid fragment is produced. The component of gamma-secretase that cleaves APP is made up of presenilin transmembrane proteins.
Mutations in the APP gene in addition to the PSEN genes which encode presenilin have been associated with early-onset AD.8 These mutations favor the formation of longer amyloid fragments which clump together into fibrils and oligomers more readily.
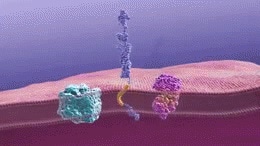
Gamma-secretase cleaves the amyloid precursor protein to generate beta-amyloid fragments. These fragments then oligomerize. Image Credit: National Institute on Aging [Public domain]
Another genetic component which has been associated with AD is the ε4 allele of the apolipoprotein E gene APOE. Possessing more copies of the ε4 allele has been linked with a higher chance of developing AD and a lower age of AD onset.9
Yet, the interaction between Aβ and apolipoprotein E (ApoE) is not well understood; ApoE is thought to regulate Aβ aggregation and clearance, but different alleles give different effects and ApoE may contribute to AD pathology via pathways which are independent of Aβ.10
Alzheimer’s Disease and the Role of Amyloid Oligomers
More recent research suggests that amyloid oligomers, rather than plaques, are neurotoxic, while the original amyloid hypothesis postulated that Aβ plaques are the toxic species that kill neurons.11
AD pathology is induced by injecting Aβ oligomers into the hippocampi of mice12 and oligomers interact with glial cells and neurons to induce oxidative stress, inflammation, and tau phosphorylation.9
Alzheimer’s Treatments Targeting Amyloid Beta
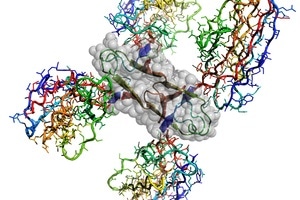
Atomic structure of the Alzheimer’s disease Amyloid-β protein caged by four shark antibodies to stop uncontrolled amyloid plaque formation. Four interacting Amyloid-β fragments are outlined by grey atomic spheres. Image Credit: CSIRO
If Aβ accumulation was the initial event that triggered neurodegeneration, then stopping this accumulation would stop AD. Some methods which could be used to address Aβ aggregation are active immunity, where people are vaccinated so they produce their own antibodies against Aβ, and passive immunization, where manufactured monoclonal antibodies are administered to patients.
In any case, these antibodies should bind to Aβ proteins and prevent them from aggregating into plaques. Crenezumab is a monoclonal antibody which binds to Aβ oligomers and monomers.
Generally, it was well tolerated, and outperformed the placebo when high doses were given to patients with very mild AD, though it did not meet the specified endpoints in a phase II trial.13
Yet, two Phase III trials were stopped when interim data showed crezenumab was not likely to meet primary endpoints. Aducanumab is another monoclonal antibody which targets aggregated Aβ. In phase II trials it was seen to decrease Aβ and slow cognitive decline.
Phase III trials were stopped after they were also established to be unlikely to meet their primary endpoints14 but after analysis of a larger data set showed promising results for patients with early-stage AD who were given high doses of the drug, Biogen now plans to file for FDA approval.
A vaccine against Aβ, AN1792, was discontinued after 6 % of participants developed meningoencephalitis (inflammation of the meninges and brain). It generated very few antibody responders and exhibited no significance between placebo groups and antibody responder after 12 months.15
Yet, a follow-up study exhibited that antibody responders had significantly less cognitive decline than the placebo recipients.16 It is feasible that treatment must be given before symptoms are shown, in order to prevent or cure AD.
Failure of certain drugs to meet clinical endpoints could be because they target the wrong biomarker, but it could also suggest that they are being administered too late to have a notable effect.
The damage to the brain may not be repaired even if amyloid plaques are cleared. In the words of a neurogeneticist at University College London, John Hardy, “The general view is that these are the right drugs, but they’re too late. It’s like taking statins when you’re having a heart attack.”17
Although, Aβ-targeted therapies have been mostly unsuccessful, leading scientists to become increasingly skeptical of the amyloid hypothesis and look at other potential pathogeneses of AD.
Evidence Against the Amyloid Hypothesis
In phase III clinical trials, bapineuzumab, solanezumab, and gantenerumab, antibodies which target Aβ, all failed to decrease cognitive decline.18,19,20 Another antibody, ponezumab, was abandoned after phase II.21
Even though there are a number of possible reasons Aβ antibodies have failed to effectively treat AD, one possibility which cannot be ignored is that Aβ accumulation does not cause neurodegeneration.
Aβ plaques happen in cognitively normal individuals and so could be a sign of normal aging.7 Without developing AD later in life, individuals with Down syndrome can develop Aβ plaques as adolescents.22 Additionally, the severity of dementia correlates more closely with NFTs than with Aβ plaques.23
The roles of APP and Aβ are still unclear. APP is upregulated following traumatic brain injury (TBI) and supplies neuroprotective effects including better cognitive and motor outcomes.24
This indicates that, rather than being the root causes of neurodegeneration, the production and aggregation of APP and Aβ may be responses to neurological damage. While the amyloid hypothesis has yet to supply an effective treatment for AD, it does not necessarily show that Aβ has no involvement in the development of the disease.
However, it does present the possibility that Aβ accumulation does not start the cascade which leads to neurodegeneration, and that other possibilities should be examined.
The Tau Hypothesis
Kosik et al. found that NFTs were made up of phosphorylated tau proteins in 1986.25 Microtubule-Associated Protein Tau (MAPT) stabilizes microtubules26 and can sustain a number of post-translational modifications including phosphorylation.
Tau dissociates from microtubules and aggregates into paired helical filaments (PHFs) and NFTs when it is hyperphosphorylated. The tau hypothesis postulates that tau tangle pathology precedes Aβ plaque formation and that tau phosphorylation and aggregation is the main cause of neurodegeneration in AD.
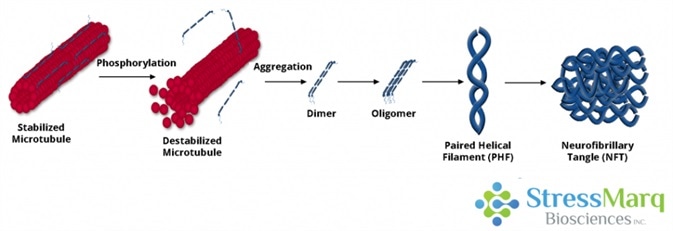
Tau dissociates from microtubules, leading to their destabilization. It then aggregates into oligomers, paired helical filaments, and ultimately neurofibrillary tangles. Image Credit: StressMarq Biosciences
Evidence for the Tau Hypothesis
Tau phosphorylation decreases its capability to promote microtubule assembly,27 causing neurodegeneration via neuronal loss and synaptic dysfunction.28 Neurofibrillary tangles can also lead to neuronal dysfunction and death.
While the amyloid hypothesis indicates that tau aggregation happens downstream of Aβ aggregation, tau tangles can be seen in the brains of patients with no Aβ pathology and very mild dementia.9 Tau pathology also correlates more closely with AD severity and progression than Aβ plaque load does.27
Whilst Aβ plaques can happen in individuals who do not experience neurodegeneration, the same cannot be said for NFTs, they are seen in frontotemporal dementia and other tauopathies. This could be because amyloid plaques are located in the extracellular space, but tau tangles happen within neurons where they can impair axonal transport severely.
Decades of focus on the amyloid hypothesis at the expense of the tau hypothesis means that tau research is generally at an earlier stage.29 Yet, tau-based strategies have exhibited some promising results29 and currently there are seven anti-tau therapies in phase II trials.30
Evidence Against the Tau Hypothesis
Sadly, a number of anti-tau therapies have also failed in clinical trials. Glycogen synthase kinase 3 beta (GSK-3β) is a protein kinase that facilitates tau phosphorylation, and therefore, is a good target for anti-tau therapies.
However, Tideglusib, which is a GSK-3β inhibitor, did not exhibit significant clinical benefit in a phase II trial.31 Methylene blue dye derivatives Trx0014 and LMTM inhibit tau aggregation and looked to slow cognitive decline in phase III trials, but the technique and claims of efficacy remain controversial.29
A Joint Mechanism
The amyloid and tau hypotheses are seen as competing, pitting “βAptists” against “Tauists”. Nonetheless, AD is multifaceted and will likely need a multifaceted approach to treatment.
There are a number of proposed theories about how tau and Aβ interact to induce AD pathology: metaphors include a toxic pas de deux,32 a trigger and a bullet,33 and a match that starts bush fires.17
Some researchers believe that tau and amyloid follow parallel but independent but mechanisms,34 but most suggest that Aβ promotes the formation of tau tangles, which in turn, damage neurons.17
There are a lot of proposed mechanisms including Aβ-induced inflammation or synaptic defects promoting tau aggregation and Aβ cross-seeding of tau.35 In addition, there is the possibility of a feedback loop where tau mediates Aβ toxicity.32
Other Possible Mechanisms
Tau tangles and amyloid plaques are not the only features observed in the AD brain; inflammation and oxidative stress also occur. The mitochondrial cascade hypothesis indicates that tau and Aβ aggregation downstream is initiated by impaired mitochondrial function, which, like AD, has a strong maternal genetic contribution.36
Herpes simplex virus type 1 (HSV1) has been linked with a heightened risk of AD in APOE-ε4 carriers, meaning viruses and other microbes may play a part in the disease.37 Acetylcholine (ACh) is a neurotransmitter that is released by motor neurons in order to activate muscles.
In AD patients it decreases in concentration and its function is weakened.38 The cholinergic hypothesis postulates that the cognitive impairment seen in AD is due to the lack of ACh.
Currently, four acetylcholinesterase inhibitors are utilized in order to treat AD. They work by inhibiting the enzymes that break down acetylcholine. These medications treat symptoms but do not cure AD. Yet, ACh dysfunction may add to AD by causing inflammation and Aβ and tau aggregation.39
The past century has seen a huge advancement in the understanding of the disease and its causes, although AD pathology is complex. A cure may not be as easy as researchers had hoped; it may be prophylactic or need a multi-pronged approach.
Recently, Bill Gates donated $ 100 million to AD research, and advancements are being made in labs around the globe. StressMarq has just launched new tau fibrils for Alzheimer’s Disease research, and supplies a variety of Aβ and tau antibodies.
References and Further Reading
- Alzheimer, A. Z. Ueber eine eigenartige Erkrankung der Hirnrinde. Allg Z Psych-Gerichtl. Med. 64, 146–148 (1907)
- Redlich, F. Uber miliare Sklerose der Hirnrinde bei seniler Atrophie. Jarbucher fur Psychiatrie und Neurologie, 1898 (1898), p. 17
- Hardy, J. A Hundred Years of Alzheimer’s Disease Research. Neuron. 52(1):3-13. (2006).
- Tomlinson, B.E., Blessed G., Roth, M. Observations on the brains of demented old people. Neurol. Sci., 11 (1970), pp. 205-242
- Mann DM. Histopathology. 1988;13(2):125–137
- Glenner, G., Wong, C. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Bioc & Biophys Res Comm. 122(6):1131-1135. (1984).
- Morris et al. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathologica Communications 2014, 2:135.
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging. 2008 Mar; 29(3):319-28.
- Jesus R. de Paula, V., Guimaraes, F.B., Diniz, B.S., Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein, or both? Dement Neuropsychol. 2009 Jul-Sep; 3(3): 188–194.
- Kanekiyo, T., Xu, H., Bu, G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. (2014). 81(4): 740-754.
- Kim, J., Basak, J.M., Holtzman, D. The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron. 2009; 63(3): 287-303.
- Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buée L (2012) Neurotoxicity and Memory Deficits Induced by Soluble Low-Molecular-Weight Amyloid-β1–42 Oligomers Are Revealed In Vivo by Using a Novel Animal Model. J Neurosci. 32:7852–7861
- Cummings, J.L. et al. A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology® 2018;0:e1-e9
- investors.biogen.com/news-releases/news-release-details/eisai-and-biogen-announce-positive-topline-results-final
- Gilman, S. et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. (2005). 64(9).
- Vellas, B. et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. (2009). 6(2): 144-51.
- nature.com/articles/d41586-018-05719-4
- Honig, L. S. et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med 2018; 378:321-330.
- Salloway, S. et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N Engl J Med 2014; 370:322-333.
- Rygiel, K. Novel strategies for Alzheimer’s disease treatment: An overview of anti-amyloid beta monoclonal antibodies. Indian J Pharmacol. (2016). 48(6): 629-636.
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry. (2018). 83(4): 311-319.
- Zigman WB, Devenny DA, Krinsky-McHale SJ, Jenkins EC, Urv TK, Wegiel J, Schupf N, Silverman W (2008) Alzheimer’s Disease in Adults with Down Syndrome. Int Rev Res Mental Retardation 36:103–145.
- Nelson, P.T. et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J Neuropathol Exp Neurol. (2012). 71(5): 362-381.
- Plummer, S. et al. The Neuroprotective Properties of the Amyloid Precursor Protein Following Traumatic Brain Injury. Aging Dis. (2016). 7(2): 163-179.
- Kosik, K.S, Joachim, C.L, and Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. (1986). 83(11): 4044-4048.
- Lindwall G, Cole RD. The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J Biol Chem. 1984 Oct 10;259(19):12241–12245.
- Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991). 82(4):239-59.
- Di, J. et al. Abnormal tau induces cognitive impairment through two different mechanisms: synaptic dysfunction and neuronal loss. Scientific Reports, 6 (2016).
- Medina, M. An Overview on the Clinical Development of Tau-Based Therapeutics. Int J Mol Sci. (2018). 19(4):1160.
- Cummings, J. et al. Alzheimer’s disease drug development pipeline: 2018. Alz & Dem: Transl Res & Clin Interventions. (2018). 4:195-214.
- Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz HJ, Calero M, Andrés MV, Gómez-Carrillo B, León T, del Ser T, ARGO investigators. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis. 2015; 45(1):75-88.
- Ittner, L. M. & Gotz, J. Amyloid-β and tau – a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. (2011). (12):67-72.
- Bloom, G.S. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. (2014). 71(4):505-8.
- Small, S.A., & Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron. (2008). 60(4): 534-542.
- Stancu, I., Vasconcelos, B., Terwel, D., & Dewachter, I. Models of β-Amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener. (2014). 9:51.
- Swerdlow, R. et al. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis. Journal of Alzheimer’s Disease. (2010). 29:S265-S279.
- Itzhaki, R.F. et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. (1997). 349(9047):241-4.
- Francis, P.T. et al. The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. (2005). 10(11 Suppl 18):6-9.
- Shen ZX (2004). “Brain cholinesterases: II. The molecular and cellular basis of Alzheimer’s disease”. Hypotheses. 63 (2): 308–21.
Acknowledgments
Produced from materials originally authored by Patricia Thomson from StressMarq Biosciences Inc.
About StressMarq Biosciences
Established in 2007, StressMarq Biosciences Inc. is a supplier of life science products that operates out of Victoria, Canada with a small, but dedicated group of scientists. Headed by our CEO and President Dr. Ariel Louwrier, StressMarq provides the research community with high-quality reagents backed with rigorous quality control data, expert scientific support, and fast international delivery.
“Discovery through partnership, Excellence through quality”
With over 7,000 products, our growth can be attributed to the continual production of cutting edge research products. Our diverse portfolio of primary antibodies, antibody conjugates, proteins, immunoassay kits and small molecules bridges across the life sciences, including products for cancer research, cardiovascular disease, cell signaling and neuroscience. To aid research worldwide, StressMarq has an extensive network of international distributors that allow us to supply reagents to over 50 countries.
In the years to come, StressMarq will continue to aid life science research by providing “Discovery through partnership, and Excellence through quality”.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.