Transthyretin (TTR) was previously known as “prealbumin” and is a protein that transports retinol and thyroxine. It is found mainly in plasma, synthesized in the liver, and makes up a big component (25 %) of cerebrospinal fluid (CSF).1 It does not cross the blood-brain barrier but can also be found in the vitreous humor of the eye.2
Transthyretin Structure
Located at the center of the molecule, transthyretin is a 55 kDa protein which is a native homotetramer with two thyroxine-binding sites in a 50 Å channel.3 Via hydrogen bonding, two monomers join edge-to-edge to form a dimer.2 Then, via hydrophobic interactions, two dimers join to form a tetramer.3
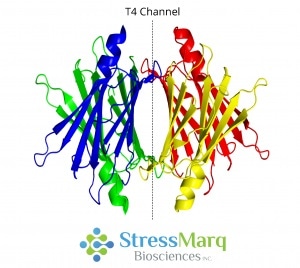
Transthyretin Tetramer with T4 Thyroxine Binding Channel. Image Credit: StressMarq Biosciences
Transthyretin in Diseases
Transthyretin can aggregate into fibrils and get deposited in the nervous system (neuropathy) and heart (cardiomyopathy). This is called transthyretin amyloidosis. Depending on whether the disease is familial and where the TTR gets deposited, there are a number of variations.
- Senile Systemic Amyloidosis (SSA) is similar to FAC, but is due to the accumulation of WT TTR not mutant TTR.4 The age of onset is typically over 60, and more than 25% of people over the age of 80 have cardiac deposition of WT TTR.5 This deposition results in heart failure.
- Familial Amyloid Cardiomyopathy (FAC) is due to the accumulation of TTR in the heart and is associated with a number of mutations.
- Familial Amyloid Polyneuropathy (FAP) is inherited in an autosomal dominant pattern and is associated with the V30M mutation. It is caused by the accumulation of TTR amyloid around peripheral nerves.2 The average age of onset is in the fourth decade of life.2
Mechanisms of TTR Aggregation
It is thought that TTR aggregation begins with the tetramer dissociating into monomers, which are prone to misfolding and self-aggregation.6 This aggregation happens in acidic conditions which are similar to those found in lysosomes.7,8 The misfolded monomers assemble into fibrils via protofibrils and oligomers.9
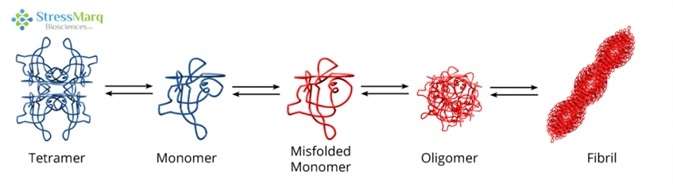
Transthyretin Misfolding and Aggregation Mechanism. Image Credit: StressMarq Biosciences
The rate-limiting step is the tetramer disassembly; so, fibril formation rates are established by tetramer dissociation rates.10 Not all monomers continue on the path to aggregation; they are also able to properly refold and combine to form dimers, which then associate to form tetramers.11
TTR Mutations and Aggregation
By studying the influences of both protective and disease-causing mutations, factors which promote TTR tetramer dissociation and monomer aggregation can be better understood.
More than 100 TTR mutations are known to be amyloidogenic.12 There is a “hot spot region” between amino acid residues 42 and 58, but amyloidogenic mutations can extend across the whole protein.13,14
Common in Swedish, Portuguese, and Japanese populations, the V30M mutation is linked with autosomal dominant familial amyloid polyneuropathy. The V30M TTR tetramer is less stable than the WT TTR tetramer, but dissociates into monomers more gradually.10
This slow dissociation rate is related to low disease penetrance which grows with age.15 V30M TTR aggregation is also affected by the rate at which unfolded monomers can refold into tetramers and dimers.
The slow rate of refolding results in the accumulation of monomers that form fibrils.16 The L55P mutation is rare, but generates aggressive amyloidosis with severe pathology. This is because the L55P mutation lowers the activation barrier for the dissociation of a tetramer to monomer, which enables dissociation under physiological conditions.17
This lowering of the activation barrier happens because the initial tetrameric state is destabilized to a bigger extent than the transition state.10 Individuals with both the T119M and V30M mutations do not develop FAP as the T119M mutation is protective against amyloidosis.18
This is echoed by the reluctance of T119M TTR to aggregate in solution when merged with V30M TTR.18 Due to an increased activation barrier that prevents tetramers from dissociating into monomers, T119M tetramers are extremely slow to dissociate.19 This activation barrier is the result of a destabilized dissociative transition state and a stabilized quaternary structure of the tetramer.20
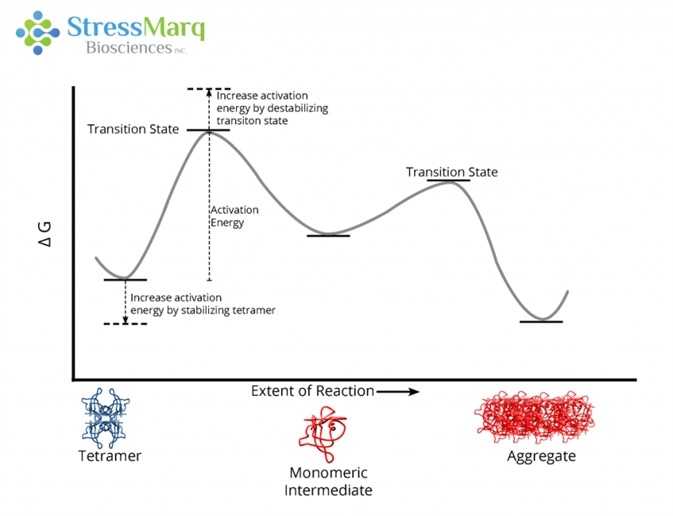
The T119M mutation prevents aggregation by increasing the activation energy required for the tetramer to dissociate into monomers. Image Credit: StressMarq Biosciences
Disease-preventing mutations, like R104H and T119M add hydrophobic residues to the thyroxine (T4)-binding cavity, which creates contact between amino acid residues of different dimers and stops dissociation.21
Throughout the human lifespan, TTR is present in the body, but does not aggregate until later in life in the absence of mutations. There are a number of aging-associated processes which could add to its aggregation, including mitochondrial dysfunction, inflammation, oxidative stress, and metabolic dysregulation.22
Transthyretin Toxicity
As with other diseases, the smaller TTR oligomers are thought to be more toxic than fibrils.2 These toxic oligomers are thought to be under 100 kDa, made up of between 6 and 10 monomers.4,23,24
These small species can bind to lipid membranes, disrupting lipid rafts and activating voltage-gated calcium channels.24 The calcium influx that ensues can induce cytotoxicity.24
Transthyretin Fibril Seeding
It is evident that ex-vivo transthyretin fibrils can recruit WT transthyretin into aggregates.25 This seeded polymerization mechanism could supply another route for TTR aggregation, and could demonstrate why in some instances, stabilizing tetramers is not enough to halt disease progression.25
Therapeutics Targeting Transthyretin
Liver transplantation is utilized to treat familial amyloid diseases by replacing mutant TTR with WT TTR, as mutant TTR is synthesized by the liver. Yet, occasionally TTR still accumulates in the heart26 and eyes.27
Diflunisal and tafamidis are TTR kinetic stabilizers which have been linked with better survival in cardiac amyloidosis patients and function by binding in the T4 binding channel.28-30

Structure of WT TTR complexed with tafamidis. Image Credit: PDB ID: 3TCT.31
Other therapeutic methods include gene silencing therapies which are used to stop the expression of amyloidogenic mutations. Some flavonoids and other small molecules inhibit TTR oligomerization,32-34 and doxycycline combined with Tauroursodeoxycholic acid (TUDCA) has been revealed to disrupt TTR fibrils in mouse models.35
Immunization with Y78F mutant TTR has also been shown to decrease TTR deposition in transgenic mice.36
Transthyretin Filaments
StressMarq supply Y78F and L55P mutant TTR monomers and filaments for researchers to examine the mechanisms of TTR aggregation and techniques to inhibit fibrillization and decrease toxicity.
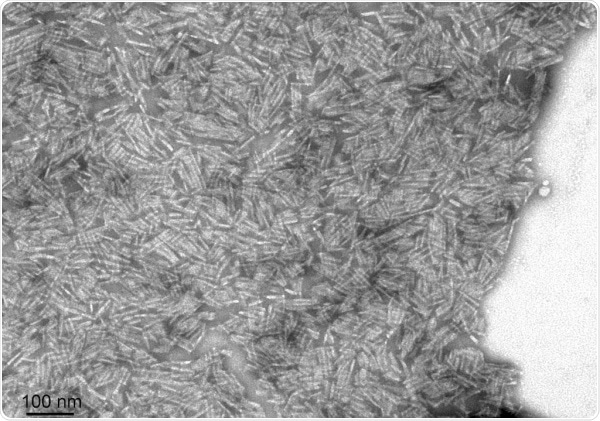
TEM of Human Recombinant Transthyretin L55P Variant Protein Filaments (SPR-464). Image Credit: StressMarq Biosciences
References and Further Reading
- Herbert, J., Wilcox, J.N., Pham, K.T., Fremeau, R.T., Jr., Zeviani, M., Dwork, A., Soprano, D.R., Makover, A., Goodman, D.S., Zimmerman, E.A., Roberts, J.L., Schon, E.A. Neurology 1986, 36(7):900-911.
- Hou, X., Aguilar, M., Small., D. Transthyretin and familial amyloidotic polyneuropathy. FEBS Journal 2007, 274:1637-1650.
- Blake, C.C., Burridge, J.M., Oatley, S.J. X-ray analysis of thyroid hormone binding to prealbumin. Biochem Soc Trans 1978, (6):1114–1118.
- Faria, T.Q., Almeida, Z.L., Cruz, P.F., Jesus, C.S.H., Castanheira, P., Brito, R.M.M. A look into amyloid formation by transthyretin: aggregation pathway and a novel kinetic model. Phys Chem Chem Phys. 2015, 17(7255):7255-7263.
- Herbert, J., Wilcox, J.N., Pham, K.T., Fremeau, R.T., Jr., Zeviani, M., Dwork, A., Soprano, D.R., Makover, A., Goodman, D. S., Zimmerman, E. A., Roberts, J.L., Schon, E.A. Transthyretin: a choroid plexus-specific transport protein in human brain. Neurology 1986, 36(7):900-911
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004, 15(1):3–16.
- Kelly, J.W., Colon, W. Partial denaturation of tranthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31:8654-8660
- Schwarzman, A.L., Tsiper, M., Wente, H., Wang, A., Vitek, M.P., Vasiliev, V., Goldgaber, D. Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid 11(1):1-9.
- Quintas A, Vaz DC, Cardoso I, Saraiva MJ, Brito RM. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem 2001, 276(29):27207–27213.
- Hammerstrom, P., Jiang, X., Hurshman, A.R., Powers, E.T., Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. PNAS. 2002, 99(4): 16427-16432.
- Jesus, C.S.H., Vaz, D.C., Saraiva, M.J.M., Vrito, R.M.M. The V30M Amyloidogenic Mutation Decreases the Rate of Refolding Kinetics of the Tetrameric Protein Transthyretin. Spectroscopy. (2012). 27(5-6):343-348.
- National Library of Medicine (US). Genetics Home Reference [Internet]. Bethesda (MD): The Library; 11 June 2019. TTR gene. [reviewed 2009 Jan; cited 20 June 2019]. Available from: ghr.nlm.nih.gov/gene/TTR.
- Saraiva, M. Transthyretin mutations in health and disease. Human Mutation 1995, 5:191-192.
- Kelly, J.W., Lansbery, P.T. Jr. A chemical approach to elucidate the mechanism of transthyretin and b-protein amyloid fibril formation. Amyloid 1994, 1:186-205
- Hellman, U., Alarcon, F., Lundgren, H.E., Suhr, O., Bonaiti-Pellie, C., Plante-Bordeneuve, V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 2008, 15(3):181-186.
- Jesus, C.S.H., Vaz, D.C., Saraiva, M.J.M., Vrito, R.M.M. The V30M Amyloidogenic Mutation Decreases the Rate of Refolding Kinetics of the Tetrameric Protein Transthyretin. Spectroscopy 2012, 27(5-6):343-348.
- Lashuel, H.A., Wurth, C., Woo, L., Kelly, J.W. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochem 1999, 38(41):13560-73.
- Coelho, T., Chorao, R., Sousa, A., Alves, I., Torres, M.F. and Saraiva, M.J.M. Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Disord. 1996, 6:27-32.
- Sun, X., Dyson, H.J., Wright, P.E. Kinetic analysis of the multistep aggregation pathway of human transthyretin. Proc Natl Acad Sci USA, 2018, 115(27):E6201-6208.
- Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science 2001; 293:2459-2461.
- Sant’Anna, R., Almeida, M.R., Varajao, N., Gallego, P., Esperante, S., Ferreira, P., Pereira-Henriques, A., Palhano, F.L., de Carvalho, M., Foguel, D., Reverter, D., Saraiva, M.J., Ventura, S. Cavity filling mutations at the thyroxine-binding site dramatically increase transthyretin stability and prevent its aggregation. Sci Rep 2017, 7:44709.
- Liao, R., Ward, J.E. Amyloid Cardiomyopathy – Disease on the Rise. Circ Res. 2017, 120(12):1865-1867.
- Reixach, N., Deechongkit, S., Jiang, X., Kelly, J. W. & Buxbaum, J. N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Natl. Acad. Sci. USA 2004, 101:2817–2822.
- Hou, X. et al. Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. Neurochem. 2007, 100:446–457.
- Saelices, L., Chung, K., Lee, J.H., Cohn, W., Whitelegge, J.P., Benson, M.D., Eisenberg, D.S. (2018). Amyloid seeding of transthyretin by ex vivo cardiac fibrils and its inhibition. PNAS 2018, 115(29):E6741-E6750.
- Liepnieks, J.J., Benson, M.D. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid, 2007. 14:277–282.
- Ohya Y, Okamoto S, Tasaki M, et al. Manifestations of transthyretin-related familial amyloidotic polyneuropathy: long-term follow-up of Japanese patients after liver transplantation. Surg Today. 2011, 41(9):1211–1218.
- Coelho, T., Maia, L. F., Martins da, S. A., Waddington, C. M., Plante-Bordeneuve, V., Lozeron, P., Suhr, O. B., Campistol, J. M., Conceicao, I. M., Schmidt, H. H., Trigo, P., Kelly, J. W., Labaudiniere, R., Chan, J., Packman, J., Wilson, A., and Grogan, D. R. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012, 79:785−792.
- Berk, J. L., Suhr, O. B., Sekijima, Y., Yamashita, T., Heneghan, M., Zeldenrust, S. R., Ando, Y., Ikeda, S., Gorevic, P., Merlini, G., Kelly, J. W., Skinner, M., Bisbee, A. B., Dyck, P. J., and Obici, L. The diflunisal trial: study accrual and drug tolerance. 2012, Amyloid 19, 37−38.
- Rosenblum, H., Castano, A., Alvarez, J., Goldsmith, J., Helmke, S., Maurer, M.S. TTR (Transthyretin) Stabilizers Are Associated With Improved Survival in Patients With TTR Cardiac Amyloidosis. Circ Heart Fail. 2018, 11(4):e004769.
- Bulawa, C.E., Connelly, S., Devit, M., Wang, L., Weigel, C., Fleming, J.A., Packman, J., Powers, E.T., Wiseman, R.L., Foss, T.R., Wilson, I.A., Kelly, J.W., Labaudiniere, R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA 2012, 109:9629-9634.
- Ferreira, N., Pereira-Henriques, A., Almeida, M.R. Transthyretin chemical chaperoning by flavonoids: Structure–activity insights towards the design of potent amyloidosis inhibitors. Biochem & Biophys Rep. 2015, 3:123-133.
- Yokoyama, T., Takaki, S., Chosa, K., Sato, T., Suico, M.A., Teranishi, Y., Shuto, T., Mizuguchi, M., Kai, H. Structural stabilization of transthyretin by a new compound, 6-benzoyl-2-hydroxy-1H-benzo[de]isoquinoline-1,3(2H)-dione. J Pharmacol Sci 2015, 129(4):240-3.
- Jono, H., Anno, T., Motoyama, K., Misumi, Y., Tasaki, M., Oshima, T., Mori, Y., Mizuguchi, M., Ueda, M., Shono, M., Obayashi, K., Arima, H., Ando, Y. Cyclodextrin, a novel therapeutic tool for suppressing amyloidogenic transthyretin misfolding in transthyretin-related amyloidosis. Biochem J 437(1):35-42.
- Cardoso, I., Martins, D., Ribeiro, T., Merlini, G., Saraiva, M.J. Synergy of combined Doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010, 8:74.
- Terazaki, H., Ando, Y., Fernandes, R., Yamamura, K., Maeda, S., Saraiva, M.J. Immunization in familial amyloidotic polyneuropathy: counteracting deposition by immunization with a Y78F TTR mutant. Lab Inv. 2006), 86:23-3
Acknowledgments
Produced from materials originally authored by Patricia Thomson from StressMarq Biosciences Inc.
About StressMarq Biosciences
Established in 2007, StressMarq Biosciences Inc. is a supplier of life science products that operates out of Victoria, Canada with a small, but dedicated group of scientists. Headed by our CEO and President Dr. Ariel Louwrier, StressMarq provides the research community with high-quality reagents backed with rigorous quality control data, expert scientific support, and fast international delivery.
“Discovery through partnership, Excellence through quality”
With over 7,000 products, our growth can be attributed to the continual production of cutting edge research products. Our diverse portfolio of primary antibodies, antibody conjugates, proteins, immunoassay kits and small molecules bridges across the life sciences, including products for cancer research, cardiovascular disease, cell signaling and neuroscience. To aid research worldwide, StressMarq has an extensive network of international distributors that allow us to supply reagents to over 50 countries.
In the years to come, StressMarq will continue to aid life science research by providing “Discovery through partnership, and Excellence through quality”.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.