Traditionally, vaccine manufacture has accounted for a huge amount of aseptic production. In recent years, there has been a larger focus on novel biologic therapies, of which the majority are administered through an injection or IV. Main pharmaceutical businesses are investing in pushing the development and release of these novel biologics. The majority of these will be producing with aseptic processing1.
When properly configured, continuous non-viable particle monitoring (NVP) offers helpful information about the variations in the aseptic environment with minute-by-minute updates of count populations and trends. This catches contamination events and therefore identifies and quantifies risks, for example, a non-zero result1.
The FDA and EU both specifically reference continuous monitoring of NVP during aseptic processing to determine real time event detection.
Main considerations when implementing an NVP system
Determine the contamination event risks to establish monitoring locations
Usually, the monitoring of fill operations occurs at three locations: (1) where sterile containers enter Grade A, (2) at the point of filling, and (3) where open and filled containers are exposed before capping. This is adequate for small, compact filling operations. However, there are potential risks that should be considered to determine if further monitoring locations are required:
- Points of human entry into the Grade A area. E.g. loading stoppers, tidying up broken vials, maintenance doors, etc.

Figure 1. NVP monitored Hopper inside clear isolation barrier. Image Credit: Beckman Coulter
- Interference with unidirectional flow. Deviation from SOPs with regards to configuring/stowing systems before sterile product exposure can be a risk. Some systems have equipment that can be higher than the sterile product throughout pre-production activities, or when reconfiguring the line.
- Reconfigurable lines. Equipment or access doors/ports which are positioned correctly for the stated process can supply an access point to the sterile product for particles.
- Moving robotics. Failed bearings or seals can create particles. Moving robotics create air turbulence which can transport these particles to the sterile product.

Figure 2. Sampling at point of line configuration. Image Credit: Beckman Coulter
- Particles from packaging components. If there is breakage or spillage of components when being loaded into the process, particles can be created that could enter the sterile product.
- Failures of power or equipment. In the brief absence of airflow, extra monitored points may be required to confirm the sterile product is not exposed to contamination throughout these events. (Note: NVP systems are able to serve the role of proving that environmental control was maintained throughout these events as it is fairly easy and affordable to guarantee continuous power for the NVP system.)
Understand the workflow
It is essential to judiciously review the media fill study and the following workflow to aid in identifying the monitoring process step and report contents required. The majority of aseptic processes are batch processing workflows, which can have several steps. For instance, when NVP monitoring in a liquid fill operation, some, or all, of these workflow steps may be implemented:
- Configuring the line. No monitoring is required during this step.
- Cleaning/sterilizing of the line. The air particle counters should remain free of ingesting liquid cleaning agents throughout this process. Airflow through the sensors should be ceased and the isokinetic sample probes capped or in some way protected from gathering caustic cleaning agents like acetic acid or bleach, which have the potential to contaminate or cause harm to the optics in the particle sensors.
- Particle sensor verification (typically called “zero counting”). Following sterilization of the environment, testing of the particle sensors should take place with absolute purge filters, or while the environment is at rest. This guarantees that any residual liquid vapor or particles gathered in the isokinetic probes during line configuration are cleaned from the sensors, and therefore guarantees false counts are not included in test data. Workers can monitor the particle counts until they reach zero, which should take place within minutes. (Note: Most contemporary aseptic processing areas will generate zero particles at 0.5 micron when they are at rest.)
- Environment “At Rest” testing. This is a distinct length sample period, for example, 36 minutes, before sterile product exposure, with the particle counters sampling. Alarms are active employing limits set at the lower limits of the “At Rest” specification for the environment. The objective is to prove the environment meets specifications before sterile product exposure. This test confirms that the environment is generating the cleanest air possible. If alarms arise, automatic retest or supervisor approval may be needed before exposing sterile products and starting the batch process.
- Batch monitoring “In Operation”. This is the stage of sterile product exposure to the environment during production activities. Alarm limits are set to sense contamination events, as discussed previously, which results in alarm levels being set below the “In Operation” limits of ISO 14644 for 0.5-micron particles. Advanced NVP systems can automatically label data gathered during this time with the Batch ID to enable the creation of batch reports for product release. Alarm acknowledgments during this time can capture “alarm reasons” by the operators to enable investigations of the root cause.
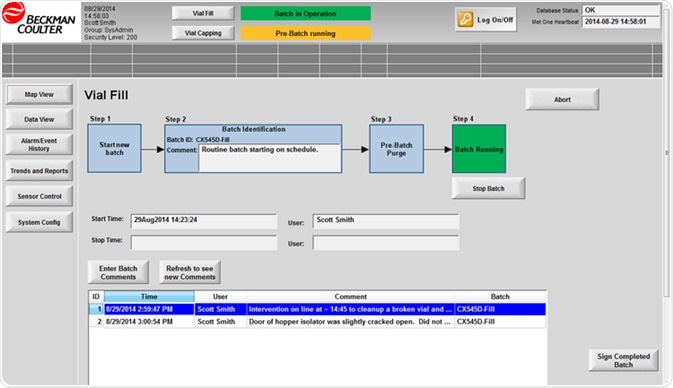
Figure 3. Batch workflow built into monitoring software. Image Credit: Beckman Coulter
- Batch pause/intervention. Some NVP systems facilitate a pause in batch data gathering during line interventions. If the line is ceased, for instance, to tidy up broken vials, and sterile product is shielded or otherwise protected. Then, the particle counters can continue sampling for QA purposes, but alarms can be deactivated and the count records omitted from the batch release report—but only until the batch processing is continued. This is only correct if sterile product is not exposed throughout the intervention. Exposed/damaged containers must be disposed of. It is well understood that a human intervention can create superior particle levels and unprotected products should be discarded. The SOP for interventions, any remedial sterilization/cleaning, and the associated risk to product, should be well defined during the media fill study.
- Bath completion. When completion of the batch is achieved, the NVP system can automatically create the batch data report, with batch alarms, for review and approval.
The monitoring process: Who will control and review it?
Previously, when NVP monitoring of the aseptic process was mostly a manual task, it was frequently completed by QA or Microbiology staff working in the aseptic area with the production staff.
In modern aseptic environments utilizing automated NVP monitoring, the majority of the monitoring process is controlled by computer. This lessens the likelihood of monitoring errors and/or removes the requirement for QA or Microbiology staff in the aseptic area during sterile product exposure.
Nowadays, monitoring can be remotely controlled from outside the aseptic area, or it can be achieved by production staff utilizing a simple, error-free, single-button process that controls the particle counters and defines the workflow.
Decreasing the number of people in the aseptic area is one of the main benefits of automated NVP monitoring systems. Similar to isolators and RABS that lessen or eradicate people from Grade A environments, automated NVP monitoring decreases the requirements for people in the Grade B environment. In some environments, a single operator in Grade B is able to support a large fill/finish operation. Process supervision is implemented from a control room, where all equipment and processes are monitored.
Consequently, the majority of contemporary NVP automated monitoring systems utilize “client and server” systems, with numerous client interfaces for different responsibilities, like completing monitoring, supervising monitoring, acknowledging alarms, or reviewing and approving batch results. The principal system software is located on a server (or VM) somewhere on the network. Consideration of client systems should be made for the following responsibilities:
- Operator. This is the person located in the Grade B aseptic area who will begin and cease monitoring, enter the batch ID, and complete interventions.

Figure 4. Operator workstation in Grade B. Image Credit: Beckman Coulter
- Alarm acknowledgment. This could be the operator in the Grade B area, or it could be a supervisor or microbiology professional located in an office or control room. Alarm notifications can be broadcast through computer clients, lights/buzzers in Grade B, or e-mail and text messages.
- Reviewer and/or Approver of monitoring results. Frequently, this is completed by Supervisors. After the product lot/batch is finished , the batch reports need to be reviewed, investigated and approved. Supervisors may also want to monitor the process in real time if alarms have taken place.
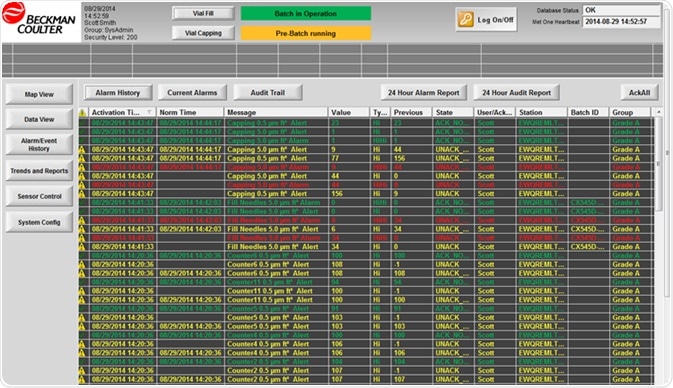
Figure 5. Supervisor alarm review screen in Internet Explorer. Image Credit: Beckman Coulter
Client computers or software will be required for every one of these tasks. Each organization functions in a slightly different way, so the number of clients/people involved will vary.
Define the audit “customers” and requirements
Government regulatory audits of the process and monitoring data are always possible. Typically, many legal entities will audit the process, and each has requirements that are a little different. Customers who buy the sterile product frequently have unique audit needs based on their internal QA standards. If there is involvement of new customers, it is sensible to check their unique needs during the design of a monitoring system.
Most aseptic producers have internal auditors and requirements that try to harmonize the variety of regulatory and customer demands. If in recent years the company needs have failed to be updated, they should be checked against the newest regulatory updates and new customer requirements:
- FDA: 2004 Guidance for Industry – Sterile Drug Products Produced by Aseptic Processing
- EU: 2009 GMP Volume 4, Annex 11 – Manufacture of Sterile Medicinal Products
- ISO 14644: Cleanrooms and Associated Controlled Environments. Note: a new and approved version is used as of November 2014
The NVP monitoring system will need to meet the demands of all auditors. There are two main areas of concern:
- Reports: The monitoring reports need to include all the necessary information: measurement data, alarm events, alarm reasons, people completing each action, reviewer and approver signatures, etc. This data needs to be accessible for any requested batch in a realistic period of time, which is normally 30 minutes to a couple of hours.
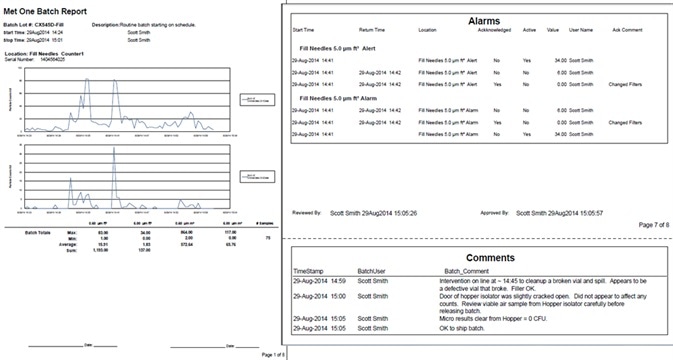
Figure 6. Sample pages from batch release report. Image Credit: Beckman Coulter
- Monitoring System: The monitoring system itself is frequently audited. Auditors would like to review alarm limits and rationale for those limits, the locations monitored, when monitoring is completed, who performs monitoring, audit trail data, data security & system access controls, validation documentation for the system, etc.
Meeting the demands of auditors can be time-consuming so it must be ensured that reports can be rapidly and easily produced from a secure database.
Additional needs
It is useful if the NVP system enables periodic review of data over a prolonged time period. Long-term reports can help guarantee that alarm limits are suitable, reflect improvements in hygiene levels, and can display repeat occurrences of the same alarm reasons, enabling possible process enhancements to advance particle control. The monitoring system should enable long-term data mining for continuous improvement efforts.
Summary
It is best practice to think about the requirements for a non-viable particle monitoring system in the early stages of designing workflow and facility. NVP monitoring is a vital part of a modern, isolated, automated, aseptic environment, and not something to be “bolted on” as an addition. Automated systems need extra time to implement and validate.
The key to applying a successful automated monitoring system is found in suitably analyzing the aseptic workflow, recognizing risks, and defining the suitable system requirements. The first step is a User Requirements Specification.
References
- Excerpts from Modern Trends in Non-Viable Particle Monitoring during Aseptic Processing: https://www.beckman.com/resources/reading-material/application-notes/modern-trends-in-non-viable-particle-monitoring
About Beckman Coulter Life Sciences
Beckman Coulter Life Sciences is dedicated to empowering discovery and scientific breakthroughs. The company’s global leadership and world-class service and support delivers sophisticated instrument systems, reagents and services to life science researchers in academic and commercial laboratories, enabling new discoveries in biology-based research and development.
A leader in centrifugation and flow cytometry, Beckman Coulter has long been an innovator in particle characterization and laboratory automation, and its products are used at the forefront of important areas of investigation, including genomics and proteomics.
Primary activity / Product lines
- Flow Cytometry
- Centrifugation
- Particle Counting and Characterization
- Liquid Handling and Robotics
- Nucleic Acid Sample Preparation
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.