A retinoblastoma is a malignant tumor of the retina, which is the thin sensory membrane at the back of the eyeball. It occurs as a result of a mutation in both copies of the RB1 gene that produces the pRB protein. This is a tumor suppressor protein, which inhibits the uncontrolled proliferation of mutant cells. When this protein is defective or absent, the abnormal cells may start to multiply in an unregulated fashion, leading to malignant transformation.
Who Should be Screened?
People at risk for retinoblastoma should be screened for the disease. Risk factors include:
- Low socioeconomic status – developing countries have a higher incidence
- Known to have a mutation in chromosome 13q
- Family history of retinoblastoma
Screening Methods
Children with a family history of retinoblastoma need to be examined by an ophthalmologist immediately after birth. These examinations should be continued for the next 5-6 years, at least, once every 2-6 months.
Parents should bring their child for ophthalmic examination if they notice the absence of the normal red pupillary reflex. This red reflection from the retina is caused by the dense network of blood vessels that supplies the sensory retina. In retinoblastoma, this normal reddish tinge is replaced by an opaque white reflection, either partially or completely. This is called leukocoria or ‘cat’s eye pupil.’ It is seen in 60% of children with leukocoria, and is the first sign of the condition in most cases.
In addition to the red reflex, the physician will check the corneal light reflex. This refers to the reflection of light off the shiny cornea when a light is shone on the eye. In physiologically normal eyes, the location of the reflected light will be identical as both eyes move together when looking at an object. In 20% of children with retinoblastoma, the corneal light reflex will be asymmetrical, showing that the eyes are misaligned. This is known as strabismus.
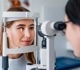
Visual function is also tested. Following this, an indirect ophthalmoscopic examination will be required to view the entire inside of the eyeball. The pupils will need to be dilated. Other eye examinations include slit lamp examination and fluorescein angiography.
In cases where the presence of a retinoblastoma is suspected, the next test may be an ultrasound or MRI scan of the eye.
A bone scan may be needed to look for bone tumors, using injected radioactive material which collects in cancerous areas of the bone.
Genetic counseling will reveal if other family members need to undergo genetic testing for the mutation in the RB1 gene. This is necessary if the patient has a heritable form of the disease.
Retinoblastomas are curable if caught early, and 90% of cases report a cure. However, recurrences and the development of other related cancers are common, especially with the heritable form of the tumor. For this reason, a lifelong follow up is required to detect these second tumors or recurrences early. Most second cancers are osteosarcomas (soft tissue cancers of the bone) or sarcomas (soft tissue cancers). In addition, these patients should not smoke because tobacco smoke is itself a potent carcinogen.
In addition, children who have a retinoblastoma in one eye need to be monitored carefully for the occurrence of the tumor in the other eye, which is common with the inherited form. This is usually seen to happen within the first 3 years after the first tumor develops, so the child should be screened with magnetic resonance imaging (MRI) scanning every six months, until the child is at least five years old.
Genetic screening through karyotyping, polymerase chain reaction (PCR) testing, or Southern blot, among other tests, may help to identify those children in affected families who have the mutation and are therefore at risk for the cancer.
References
Further Reading
Last Updated: Jan 2, 2023
 World’s largest eye imaging and data resource expands across the UK
World’s largest eye imaging and data resource expands across the UK