Sickle cell disease is an inherited genetic condition that involves defects in the shape and function of hemoglobin in the blood. This increases the likelihood of blockages in the blood vessels and disrupted blood flow, which can result in serious complications.
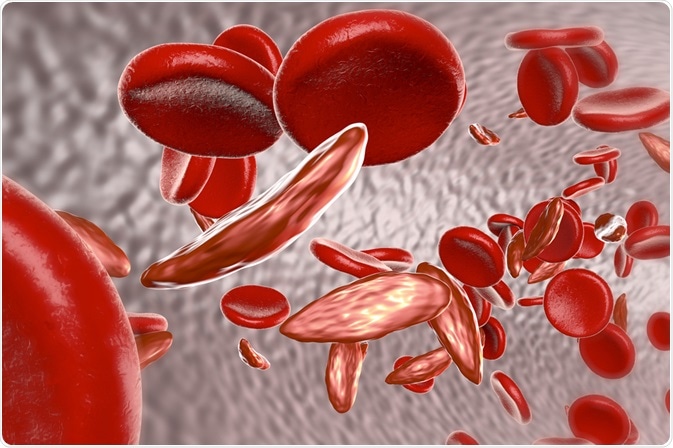
Image Credit: Kateryna Kon / Shutterstock.com
The role of hemoglobin
Hemoglobin is an essential cellular component of the red blood cells that play the role of transporting oxygen in the blood to the bodily tissues where it is required.
Sickle hemoglobin differs in physical shape from normal hemoglobin, with a curved sickle-shape rather than flat-disc-shaped cells. The shape alters the properties of the cells, causing them to become more rigid and less flexible. As a result, the cells are more likely to hemolyze and cause blockages in the blood vessels that disrupt the flow of blood.
Genetic mutation
The specific genetic mutation that results in sickle hemoglobin involves a substitution of thymine for adenine (from GAG to GTG) on the sixth codon of the genetic sequence. This leads to the coding of valine rather than glutamate on the sixth position of the hemoglobin beta chain.
This genetic alteration changes the physical properties of the hemoglobin in cells, changing their shape to the characteristic sickle shape, as well as their physical properties, such as solubility and stability. It is these properties that account for changes in function and the common complications of the disease.
Genetic inheritance pattern
Sickle cell disease is an inherited condition that follows an autosomal recessive pattern. This means that males and females are affected equally and both parents must carry the genetic mutation, even if they are asymptomatic, for the child to be affected.
When both parents carry a single gene mutation, known as the sickle cell trait, there is a 25% chance that the disease will develop in their offspring, a 25% chance that the child will be unaffected and a 50% that they will possess the genetic mutation as an asymptomatic carrier.
Pathology
When there is insufficient oxygen in the vascular system, sickle hemoglobin becomes considerably more insoluble, increasing the polymer formation in the blood and its overall viscosity. This leads to the formation of tactoids, which are a gel-like form of hemoglobin, that exists in equilibrium with its ordinary soluble state.
The proportion of each type depends on:
- Oxygen presence: more oxygen supports the prevalence of the liquid state
- Sickle hemoglobin concentration: more HbS supports gel-like state
- Other hemoglobins: Normal adult and fetal Hb support liquid state
Over time, the membrane of the cells becomes permanently damaged, leading cells to permanently stay in the bi-concave sickle shape, even when the blood is exposed to sufficient levels of oxygen once again.
Complications
Initial symptoms of sickle cell disease tend to arise in young children between the ages of approximately six months to one year old, as the high concentration of fetal hemoglobin plays a protective role before this time. There are three main complications that can arise in children prior to their diagnosis, which include sickle cell crisis, anemia, and multiple organ damage.
Sickle cell crises may result due to the increased viscosity of the blood and the formation of blockages in the blood vessels. When the rigid cells group together, they can disrupt the flow of oxygen and restrict the supply to tissues that require oxygenation. This results in sudden and severe pain, known as sickle cell crisis, which usually requires medical management.
Anemia can present due to hemolysis of the red blood cells with sickle hemoglobin in the spleen. As a result, the red blood cells have a shorter lifespan than normal, thus allowing for hemolytic anemia to potentially develop.
Multiple organ damage can occur to patients with sickle hemoglobin over an extended period of time. This may affect the heart, skeleton, spleen, brain, eyes, lungs, kidneys, penis, and skin.
References
Further Reading
Last Updated: Mar 27, 2021
 New partnership aims to expand global access to sickle cell gene therapy
New partnership aims to expand global access to sickle cell gene therapy