Epigenetics refers to the study of heritable changes that occur in gene expression. Such changes are induced by non-genetic mechanisms, without causing any change in the DNA sequence or gene structure.
Normally, a cell’s epigenetic state develops during the cellular development and differentiation of an organism, and epigenetic changes are associated with cellular reprogramming. Epigenetic mechanisms could also contribute to the integration of environmental reactions right at the cellular level and therefore, can possibly play a critical role in the development of certain diseases.
Growth factors, transcription factors, and even hormones are involved in the epigenetic regulation of gene activity, but this process is complicated and has not been understood fully. Epigenetic processes involve the alteration of chromatin.
Epigenetic events possibly involve covalent modifications of RNA, DNA, histones, in addition to micro-RNA mediation, chromatin remodeling, and other modifications to chromatin structure. As such, protein families have been detected through which these modification events are regulated.
“Writers" such as DNA methyl-transferases, histone kinases, histone methyltransfersases, and histone acetyl-transferases perform the chemical modifications. More than 80 different writer proteins have been detected so far. The marks act as docking modules for “readers" such as proteins, which identify particular acetyated or methylated lysine marks on histones.
More than 300 different reader proteins have been detected to date. Such marks are removed by “erasers" such as histone demethylases and histone deacetylases. Approximately 50 erasers have been detected so far.
Epigenetics research undertaken in the past was mainly centered on single domain-single mark relationships, and has since advanced to investigating various domain-multiple mark interactions as well as the complicated signaling and associations among the epigenetic network.
This means numerous proteins are involved in epigenetic regulation and serve as promising targets for drug discovery. The modulation of epigenetic mechanisms is applicable to many diseases, such as cancers, inflammation, metabolic disorders, viral infections, and central nervous system diseases.
Characterization of interactions with chromatin
To define the specificity and relative binding affinity for chromatin-binding proteins, it is essential to study the purified module with preferred activity, communicating with its histone mark, and eventually adhering to a histone peptide mono-, di- or triacetylated at particular position(s).
ELISA, chromatin immunoprecipitation (CHiP) [52], gel-shift assays, and histone peptide bead-based pull-down assays [45], constitute in vitro biochemical assays. Certain types of assays can be performed with nuclear extracts, while purified proteins are required in others.
In this regard, high-throughput assays to support screening, as well as compound profiling, have become more advanced as targets involved in reading, writing, and erasing histone methylation are relegated to drug discovery pipelines.
Artificial histone peptide surrogates are typically employed in high-throughput histone binding screening assays like AlphaScreen common high-throughput® [33,76], SPOT assays [48,51], and AlphaLISA® [83]. Differential scanning fluorimetry (DSF) is a standard high-throughput assay used for screening drugs and proteins.
Here, drug binding is evaluated by a change in the thermal stability of proteins. Such high-throughput, biochemical screening assays provide the logical amount of qualitative data regarding the specificity and the corresponding binding affinity by rank ordering.
However, it is to be noted that qualitative biochemical assays meant for histone-binding proteins pose several challenges with regard to nonspecific binding, artifacts, reproducibility, reliability, and also the possible false negative or false positive results.
However, the above-mentioned biochemical and screening assays fail to give precise KD values. SPOT assays provide a relative affinity depending on the spot intensity, whereas AlphaScreen® assays provide IC50 values that are related to but not the same as the KD values.
It is important to ensure that at least a single biophysical binding assay is used to determine KD, for example, isothermal titration calorimetry (ITC), microscale thermophoresis (MST), fluorescence polarization, or surface plasmon resonance (SPR).
Biophysical binding assays give quantitative KD measurements and are orthogonal to major binding screens. Such assays help verify the preliminary screening data and identify false negatives, false positives, and detection limits that are intrinsic to the original assay.
In addition, biophysical assays offer data beyond KD, such as binding stoichiometry, binding thermodynamics, and binding kinetics. Orthogonal assays generally provide these measurements, offering a better understanding of the binding mechanism, the selectivity and specificity of the interaction, and the structure of the active binding domain. Structural studies like nuclear magnetic resonance (NMR) and X-ray crystallography provide additional validation and characterization.
This article describes the features of the ITC assay and shows how ITC can be used to determine the KD of binding events that involve chromatin and epigenetic proteins. Case studies are also presented, demonstrating how the binding affinity and other similar parameters rendered by ITC are used to define certain interactions and thus design inhibitors.
Isothermal titration calorimetry
The latest ITC instruments can determine KD values in the millimolar to the single-digit nanomolar range. They can also be used to measure binding stoichiometry and binding enthalpy/entropy. ITC is a sensitive, label-free, versatile method and is used to define binding interactions that involve lipids, proteins, and nucleic acids.
Therefore, it has been established as the “gold standard” technique in the field of life sciences for studying binding processes. ITC can be used with various materials in a wide range of buffers. It is a biophysical assay and has been featured in many citations of scientific journals. Figure 1 show the operation of ITC.
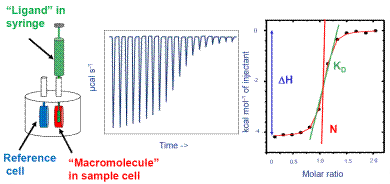
Figure 1. How ITC works. Left: Schematic of ITC cells and injection syringe. Center: Representative raw ITC data from titration experiment. Right: Representative binding isotherm from ITC experiment, fit to one set of sites binding model.
The ITC system is specifically programmed to administer some amount of ligand into the macromolecule solution at regular intervals. The mixture is continuously stirred all through the experiment because the injection syringe is provided with a paddle in the base.
Several ITC instruments are provided by Malvern Panalytical which include MicroCal iTC200 and MicroCal PEAQ-ITC. These instruments determine the temperature variation between the sample cells and the reference, and modify the thermal power compensation to bring back the temperature variation to zero.
The power compensation directly relates to the binding heat. In Figure 1, the center image shows a representation of the raw output of the ITC experiment. Here, the binding heat is exothermic and individual peaks donate the heat change promoted by the administration of ligand. The initial peaks are large, as most of the injected ligand was bound to the macromolecule.
When the ligand saturates the macromolecule, less ligand will bind together with additional injections, thereby producing less amount of heat. Towards the conclusion of the experiment, only very little ligand is bound and a small amount of heat is produced because of the heat of dilution.
On uploading the raw ITC data in the ITC data analysis software, an integration baseline is created automatically, and the region of individual peaks is measured and plotted as a function of the molar ratio of ligand to the macromolecule present in the cell. This is depicted on the right side of Figure 1.
Each peak is represented by the black dots. The red curved line represents the optimum fit to a binding algorithm from which the interaction enthalpy, the affinity, and the stoichiometry are determined.
The characterization of binding entropy and enthalpy offers a better understanding of the binding mechanism, including hydrophobic effects, hydrogen bonding, and conformational changes that take place during binding. Affinity and thermodynamics are used to elucidate the structural and specificity data associated with the binding.
ITC has become a recognized binding assay for proteins involved in epigenetic regulation of gene activity. Many historic studies describing the use of ITC for the detection and characterization of interactions between histones and chromatin reader proteins have been published [30,31,38]. ITC is used as a main binding assay and also as a validation assay following initial biochemical screening assays:
- As a universal detection system, ITC determines the heat change related to binding. In other words, native proteins can be employed, and there is no need to perform tagging, labeling, marking, or other modifications
- ITC determines KD from millimolar to nanomolar values, which are the usual range of epigenetic protein binding
- As a true in-solution technique, ITC eliminates immobilization and thus the issues related to potential stearic or orientation; it also eliminates non-specific binding to other materials such as beads
- ITC also removes the need for fluorescence measurements, and therefore it is not sensitive to possible artifacts in fluorescence-based assays
- ITC needs minimal assay development and does not require specific antibodies or reagents
- ITC can be carried out in any type of biological buffer, including those consisting of standard additives like detergents, salts, cofactors, reducing agents, and so on
- In addition to KD, ITC provides data-rich results. It determines binding stoichiometry and thermodynamics that are essential in drug discovery, structure-activity relationships, and structural biology
- As ITC has no molecular weight limitations, it can be employed with any molecular size to assess interactions between proteins, between proteins and small molecules, and between proteins and nucleic acids, and also those of large multi-protein complexes
Contemporary instruments, which are more sensitive and employ less protein as opposed to early ITCs, have been developed, due to improvements in the software and design. Such improvements have made the method more cost-effective and easier to use. ITC is also a non-destructive method, which makes it easy to recover the protein following the experiment. ITC instruments in automated formats are also available for high throughput.
Characterization of protein modules binding to nucleic acids
ITC determines the binding between chromatin’s nucleic acid component and protein domains. In a recent article, mammalian 5-methylcytosine (5mC) oxidase Tet3 was analyzed thoroughly, and the full-length isoform comprising an N-terminal CXXC domain (Tet3FL) [35] was subsequently characterized. The CXXC domain was found to be an epigenetic reader protein.
Several CcaCG-interacting mTet3-CXXC residues were selected for mutagenesis, and ITC was used to determine their impact on mTet3’s binding affinities toward CcaCG and CCG DNA. It was seen that H81 mutation to alanine removed the binding of the CXXC domain to CcaCG and CCG DNA, agreeing well with the structural observations made before.
Mutation of K88A also resulted in >27 times weaker binding toward CcaCG DNA but merely 3 times weaker binding toward CCG. Such observations underscore the fact that residue K88 contributes to particular recognition between CcaCG DNA and mTet3-CXXC.
Characterization of protein modules binding to substrate/cofactor
Several journal articles are available which cite the application of ITC to observe the interaction of a histone acetyltransferase binding its substrate - such as histone methyltransferase binding to the cofactor S-adenosyl methionine [15,79] or acetyl-coenzyme A or coenzyme A [1]
Characterization of protein modules binding to histone peptides
Researchers often use ITC to define a reader protein bound to histone peptides. Bromodomains (BRDs) are a primary class of reader proteins, with more than 60 human proteins comprising at least a single BRD module. BRDs mainly detect ε-N-lysine acetylation (Kac) motifs on histones. Numerous research articles use ITC data to examine BRD interactions [29,31,64].
In one study [36], MicroCal Auto-iTC200 was used to achieve thermodynamics and binding affinities of H3C4me3, histone H3K4me3, and H3G4 peptides bound to five reader proteins, which detect H3K4me3.
The five reader pockets were found to be different in the aromatic cage composition and design, permitting analysis of the particular effect of separate components of the aromatic cage on binding variations. A stoichiometry of 1 was found on all ITC binding assays. Comparative ITC experiments for the relationships of H3C4me3 and H3K4me3 demonstrated that:
- H3K4me3 bound 2 to 33 times more strongly as opposed to the neutral H3C4me3 bound to four out of five reader proteins containing Trp as part of the aromatic cage (Figures 2 and 3)
- When compared to the association of the neutral Cme3 group to the aromatic cage, association of the Kme3 side chain with the same aromatic cage was found to be more favorable in enthalpy (Figure 3)
- When compared to the association of the Cme3 group to the aromatic pocket, association of the Kme3 side chain was found to be less favorable in entropy (Figure 3)
Together with structural and computational results, ITC binding and thermodynamics data showed the presence of positive cation–pi interactions.
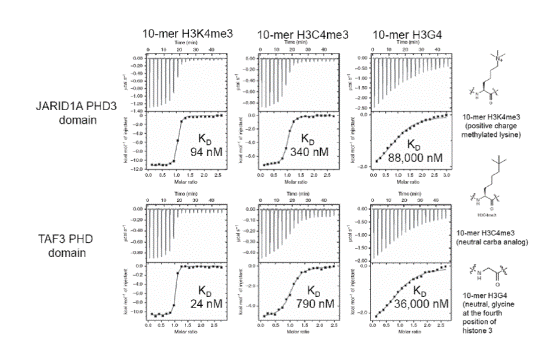
Figure 2. ITC data for JARIDA1A PHD3 reader domain (top row) and TAF3 PHD reader domain (bottom row) binding to histones H3K4me3 (left), H3C4me3 (middle) and H3G4 (right). Top of each ITC titration is raw data, bottom is binding isotherm fitted to one sets of site model. KD values are inset. Structures of the histones are on the far right. Reprinted with permission [36]
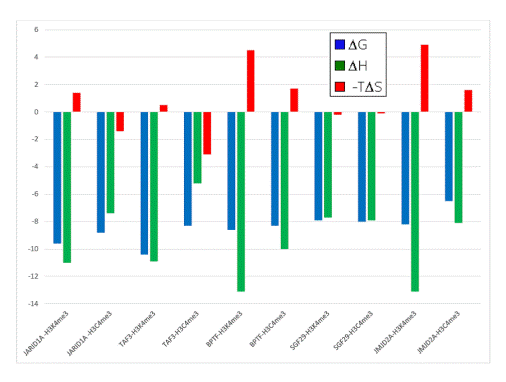
Figure 3. Thermodynamic data from ITC experiments showing the binding of five reader protein domains to H3K4me3 or H3C4me3. Blue: ∆G; green: ∆H; red: -T∆S. Negative values are “favorable” processes, positive values are “unfavorable.” Data adapted with permission [36]
The study provides both hypothetical and experimental proof that reader proteins mainly detect trimethyllysine through a combination of favorable cation–pi interactions, and contribute to the release of water molecules owing to the association with the trimethyllysine side chain. This data holds implications in practical drug design by targeting the aromatic cage of trimethyllysine readers.
Epigenetics, drug discovery, and ITC
The modulation of epigenetic mechanisms through chromatin-binding proteins is applicable to various human diseases. Epigenetic erasers, readers, and writers can be regarded as prospective druggable targets.
Methylation and acetylation are well-defined, and a better insight into the mechanisms of other modifications may lead to drug discovery in the future. During drug discovery, ITC method is used for lead optimization, hit validation, and mechanism of action studies. ITC’s stoichiometry, KD, and thermodynamics can be used to define drug-target binding and shift chemical hits into leads [20,26,40].
Given that the target protein shares structural domains with other types of proteins in the same epigenetic target family, development of tiny molecule inhibitors against a particular epigenetic domain is difficult, predominately for reader proteins such as BRDs.
In vivo studies demonstrated that the BRD4 fusion oncoprotein from chromatin was displaced by a cell-permeable small molecule JQ1, leading to specific anti-proliferative effects and squamous differentiation in xenograft models and BRD4-dependent cell lines. The resultant data provided proof of concept for protein-protein interactions of epigenetic readers and offered a unique scaffold for designing chemical probes for the BRD family.[4]
ITC along with probe screening assays/bromodomain inhibitor such as DSF and AlphaScreen® was used in other studies [9,16,21,37,47,56,57,65,82]. It was even used to determine KD of inhibitors and probes to other epigenetic protein families like MBT [25], histone deacetylase [68,69], WD40 [66], histone lysine methyltransferase [17,42,44], and chromodomains [67,72].
In one study, James et al. (2013) [32] examined possible inhibitors of the histone methyl-lysine (Kme) reader protein, L3MBTL3. Using AlphaScreen® assay, affinities for tiny molecule compounds bound to L3MBTL3 were established, and an orthogonal time resolved fluorescence resonance energy transfer (TR-FRET) assay was used to validate these binding trends.
The MicroCal Auto-ITC200 was also used to validate the binding affinity of the target compounds. The combination of ITC, TR-FRET, and AlphaScreen® provided confidence in better understanding the SAR trends.
Since compounds 2 and 56 showed potential as more selective L3MBTL3 inhibitors, ITC was used to further measure the selectivity level, comparing binding to two identical proteins, as shown in Table 1.
The combination of structural and ITC data indicates that the selectivity of L3MBTL3 inhibitors can be improved by modifying the amines that are respectively bound to the first and second MBT domain in the dimeric binding mode.
Table 1. Results of orthogonal analyses for binding affinities of small molecule compounds to L3MBTL3 and L3MBTL1. Data adapted with permission [32]
|
|
Compound 5
|
Compound 1
|
Compound 2
|
Compound 56
|
|
L3MBTL3 binding by AlphaScreen® IC50 (μM)
|
0.071
|
0.064
|
0.17
|
0.13
|
|
L3MBTL1 binding by AlphaScreen® IC50 (μM)
|
2.9
|
2.3
|
>10
|
9.6
|
|
L3MBTL3/ L3MBTL1 selectivity by AlphaScreen®
|
41
|
35.4
|
>59
|
74
|
|
L3MBTL3 binding by ITC KD (μM)
|
0.38
|
0.12
|
0.47
|
0.35
|
|
L3MBTL1 binding by ITC KD (μM)
|
6.2
|
9.4
|
0.68
|
132
|
|
L3MBTL3/ L3MBTL1 selectivity by ITC
|
16
|
78
|
145
|
380
|
Conclusion
The combination of biophysical screening and ITC’s quantitative KD values assisted in the identification and characterization of epigenetic protein interactions with chromatin. This combination has been shown to be a well-proven technique for assessing binding selectivity and specificity.
Using the right chain of histone peptides and mutant and wild-type proteins of the same epigenetic family, ITC can easily establish the contribution of each modification or residue to the overall binding affinity for a more complicated substrate.
By using the combination of stoichiometry, thermodynamic and binding data obtained from ITC, X-ray crystallography, NMR, in vivo, and other similar structural studies will provide a better insight into these complex interactions. Subsequently, SAR, ITC, biochemical screening, and in vivo assays can be employed in logical drug design to generate inhibitors that are relevant to writers, readers, and eraser targets.
References
- Albaugh, B.N., Kolonko, E.M., and Denu, J.M. Biochemistry 49, 6375-6385 (2010) doi:10.1021/bi100381y.
- Baud., M.G.J., Lin-Shiao, E., Cardote, T., et al. Science 346, 638-641 (2014) doi:10.1126/science.1249830.
- Ciceri, P., Müller, S., O’Mahony, A., et al. Nat. Chem. Biol.10, 305-312 (2014) doi:10.1038/nchembio.1471.
- Eram, M.S., Bustos, S.P., Lima-Fernandes, E., et al. J. Biol. Chem. 289, 12177-12188 (2014) doi:10.1074/jbc.M113.523183.
- Federov, O., Castex, J., Tallant, C., et al. Sci. Adv. 1, e150072313 (2015) doi:10.1126/sciadv.1500723.
- Ferguson, A. D., Larsen, N. A., Tina Howard, T., et al. Structure 19, 1262-1273 (2011) doi:10.1016/j.str.2011.06.011.
- Freire, E. Drug Disc.Today 13, 869-874 (2008) doi:10.1016/j.drudis.2008.07.005.
- Gacias, M., Gerona-Navarro, G., Plotnikov, A. N., et al. Chem. Biol. 21, 841-854 (2014) doi:10.1016/j.chembiol.2014.05.009.
- Herold, J. M., Wigle, T. J., Norris, J. L., et al. J. Med. Chem. 54, 2504-2511 (2011) doi:10.1021/jm200045v.
- Holdgate, G. Meth. Mol. Biol. 572, 101-133 (2010) doi:10.1007/978-1-60761-244-5_7,
- Itzen, F., Greifenberg, A., Bosken, C.A., and Geyer, M. Nucl. Acids Res. 42, 7577-7590 (2014) doi:10.1093/nar/gku449.
- Jacobson, R.H., Ladurner, A.G., King, D.S., and Tjian, R. Science 288, 1422-1425 (2000) doi:10.1126/science.288.5470.1422.
- James, L. I., Korboukh, V. K., Liubov Krichevsky, L. et al. J. Med. Chem. 56, 7358-7371 (2013) doi:10.1021/jm400919p.
- Janzen, W. P., Wigle, T. J., Jin, J., and Frye, S. V. Drug Discov. Today Technol. 7, e59-e68 (2010) doi:10.1016/j.ddtec.2010.07.004.
- Jin, S.G., Zhang, Z.M., Dunell, T.L., et al. Cell Reports 14, 493-505 (2016) doi:10.1016/j.celrep.2015.12.044.
- Kamps, J.J.A.G, Huang, J., Poater, J., et al. Nat. Commun. 6, 8911 (2015) doi:10.1038/ncomms9911.
- Kempen, H. J., Bellus, D., Fedorov, O., et al. Lipid Insights 6, 47-54 (2013) doi:10.4137/LPI.S13258.
- Ladbury, J. E., Klebe, G., and Freire, E. Nat. Reviews Drug Disc. 9, 23-27 (2010) doi:10.1038/nrd3054.
- Liu, F., Chen, X., Allali-Hassani, A., et al. J. Med. Chem. 53, 5844-5857 (2010) doi:10.1021/jm100478y.
- Ma, A., Yu, W., Li, F., et al. J. Med. Chem. 57, 6822−6833 (2014) doi:10.1021/jm500871s.
- Malecek, K., and Ruhenburg, A. Methods Enyzymol. 512, 187-220 (2012) doi:10.1016/B978-0-12-391940-3.00009-3.
- McLure, K. G., Gesner, E. M., Tsujikawa, L. et al. PLoS One 8, e83190 (2013) doi:10.1371/journal.pone.0083190.
- Muller, M. M. and Muir, T. W. Chem. Rev. 115, 2296-2349 (2015) doi:10.1021/cr5003529.
- Nady, N. Min, J., Kareta, M. S. et al. Trends Biochem. Sci. 33, 305-313 (2008) doi:10.1016/j.tibs.2008.04.014.
- Nelson, J. D., Denisenko, O., and Bomsztyk, K. Nat. Prot. 1, 179-185 (2006) doi:10.1038/nprot.2006.27.
- Pande, V. J. Med. Chem. 59, 1299-1307 (2016) doi:10.1021/acs.jmedchem.5b01507.
- Picaud, S., Strocchia, M., Terracciano, S. et al. J. Med. Chem. 58, 2718-2736 (2015) doi:10.1021/jm501893k.
- Picaud, S., Wells, C., Felletar, I., et al. Proc. Natl, Acad. Sci. 110, 19754-19759 (2013) doi:10.1073/pnas.1310658110.
- Ruthenburg, A.J., Li, H., Milne,T.A., et al. Cell 145, 692-706 (2011) doi:10.1016/j.cell.2011.03.053.
- Senisterra, G., Wu, H., Allala-Hassani, A., et al. Biochem. J. 449, 151-159 (2013) doi:10.1042/BJ20121280.
- Simhadri, C., Daze, K. D., Douglas, S. F. et al. J. Med. Chem. 57, 2874−2883 (2014) doi:10.1021/jm401487x.
- Singh, R. K., Lall, N., Travis S. Leedahl, T. S., et al. Biochemistry 52, 8139-8148 (2013) doi:10.1021/bi400740x.
- Singh, R. K., Suzuki, T., Mandal, T., et al. Biochemistry 53, 7445-7458 (2014) doi:10.1021/bi500711x.
- Stuckey, J. I., Dickson, B. M., Cheng, N., et al. Nat. Chem Biol. 12, 180-187 (2016) doi:10.1038/nchembio.2007.
- Wagner, E. K., Albaugh, B. N., and Denu, J. M. Methods Enzymol. 512, 161-185 (2012) doi:10.1016/B978-0-12-391940-3.00008-1.
- Wu, H., Zeng, H., Dong, A., et al. PLoS One 8, e83737 (2013) doi:10.1371/journal.pone.0083737.
- Zengerle, M., Chan, K. H., and Ciulli, A. ACS Chem. Biol. 10, 1770-1777 (2015) doi:10.1021/acschembio.5b00216
- Zhan, Y., Kost-Alimova, M., Shi, X., et al. Epigenetics & Chromatin 8, 37-56 (2015) doi:10.1186/s13072-015-0026-4.
About Malvern Panalytical

Malvern Panalytical provides the materials and biophysical characterization technology and expertise that enable scientists and engineers to understand and control the properties of dispersed systems.
These systems range from proteins and polymers in solution, particle and nanoparticle suspensions and emulsions, through to sprays and aerosols, industrial bulk powders and high concentration slurries.
Used at all stages of research, development and manufacturing, Malvern Panalytical’s materials characterization instruments provide critical information that helps accelerate research and product development, enhance and maintain product quality and optimize process efficiency.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.