Choroideremia (CHM) is a rare genetic eye disease that affects the retina, which is the area at the back of the eye that processes all we see into signals that are sent to the brain via the optic nerve.
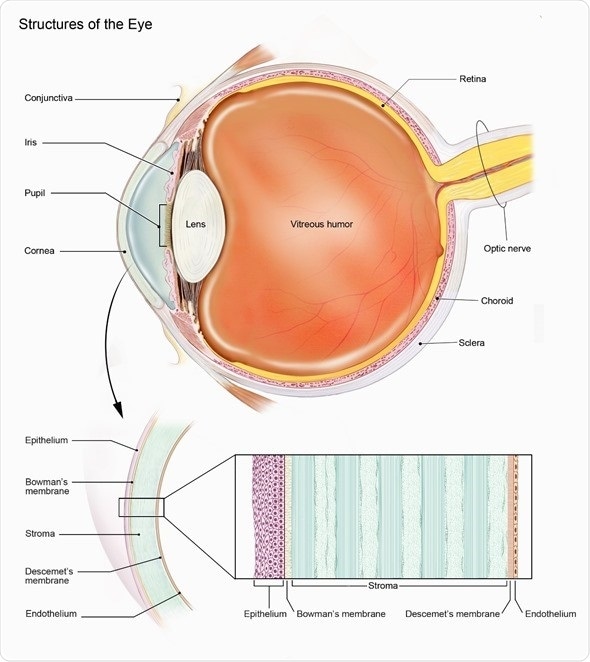
Choroideremia affects about 1 in 50,000 individuals and is caused by a genetic defect in one single gene called the CHM gene, which is located on the X-chromosome.
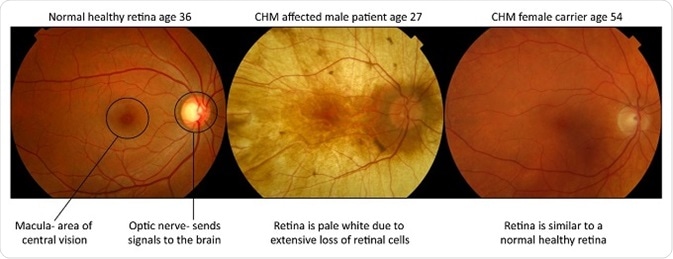
Male patients manifest the severe blinding form of the disease and suffer from a progressive retinal degeneration with obliteration of the light sensing cells (photoreceptors), their support cells (the retinal pigment epithelium) and part of the blood supply to these retinal layers (the choroid).
CRF Funded Researcher Dr. Mariya Moosajee
The healthy orange retina is lost and the pale white sclera (tough white coat of the eye) can be seen when you look into the eye. Females are carriers and largely unaffected, however they can develop mild symptoms of visual impairment, such as night blindness, in more in later stages of life.
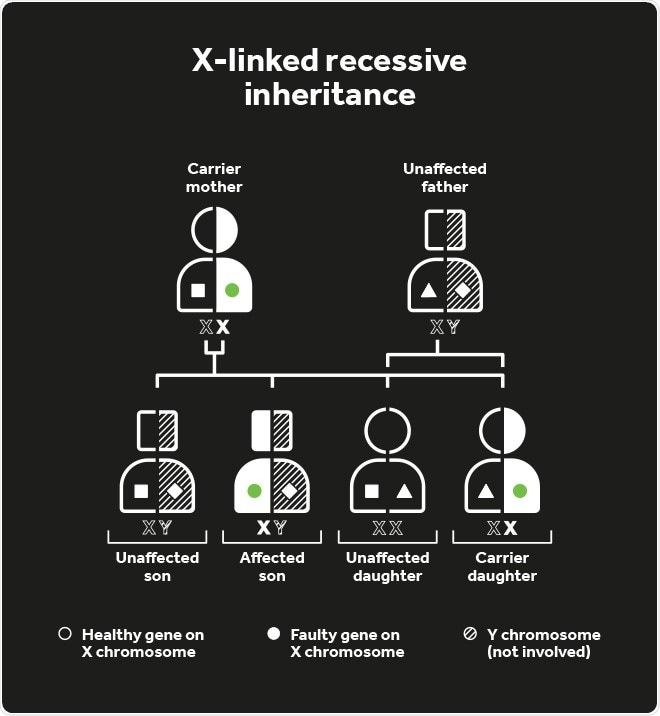
Choroideremia inheritance
An affected parent has a 50% chance of passing the defective CHM gene to their children, although as it is positioned on the X-chromosome, affected males can only pass the disease gene to their daughters (so they become carriers) and not their sons. Female carriers have a 50% risk of passing the disease to their sons.
The CHM gene and how it works
The CHM gene encodes a protein called Rab Escort Protein-1 (REP1). This protein works in all our cells by helping to shuttle other small proteins (called Rabs) to their correct destination so they can undertake their role, such as allowing nutrients to pass across cells and removal of waste products.
If these small proteins cannot be transported to their correct location to fulfill their job, the cells can starve and waste products build up causing damage and cell death. Humans are fortunate to have a second copy of REP1 called Rab Escort Protein-2 (REP2), which works well to transport Rabs in all the cells of our body except for the retina.
The photoreceptors and retinal pigment epithelium have a subset of Rab proteins that prefer to be escorted by REP1 over REP2. Therefore, in the presence of a defective CHM gene with lack of REP1, there is a buildup of Rabs that are not able to function appropriately, and this leads to a retinal-specific disease that causes blindness in an otherwise fit and healthy individual.
Choroideremia symptoms and disease progression
In affected male patients, the first symptom of CHM is night blindness, which is most commonly noticed in early childhood from as early as five or six years of age.
The disease continues to progress with loss of the peripheral field of vision, this occurs at a relative fast rate in late adolescence and into the early twenties, leaving the patient with only a small area of central “tunnel” vision.
The rate of decline lessens but the retinal degeneration continues to progress slowly over the next few decades, patients lose depth perception, color perception and finally loss of central vision with complete blindness into their fifties and sixties.
Both the rate of disease progression and the degree of visual loss are variable among those affected, even within the same family.
Female carriers are usually unaffected, but some do experience symptoms of night blindness later in life with some patchy loss in vision. This occurs because despite women having two X-chromosomes, one is switched off in every cell to ensure the correct amount of gene product (protein) is being produced.
This process is random, called X-inactivation or lyonization, and means if the healthy X-chromosome is inactivated, then the X-chromosome carrying the defective CHM gene will prevent normal function in that particular retinal cell leading to disease in a patchy distribution.
Diagnosis of choroideremia
A diagnosis can be made by an ophthalmologist with a specialist interest in genetic eye disease.
They will have the expertise to interpret the patient history, signs from the clinical examination and investigations such as electrophysiology, fundus autofluorescence and optical coherence tomography. But ultimately, a genetic test to screen the CHM gene for mutations is required to make a definitive diagnosis.
There have been rare cases of patients with a retinal appearance similar to CHM, but upon genetic testing have shown mutations in different genes known to cause other retinal dystrophies, such as RPE65.
Another genetic condition called gyrate atrophy, which is an inborn error of metabolism, displays similar clinical features to CHM. It is caused by mutations in the ornithine ketoacid aminotransferase (OAT) gene found on chromosome 10.
Patients show an increase in plasma levels of ornithine, however, reduction of the amino acid arginine in the diet prevents disease progression. This highlights the need to see a specialist so that CHM can be diagnosed correctly.
Treatments options
There is currently no cure or effective treatment for CHM. Much research is underway to seek a viable and sustainable therapy and there are a number of promising gene-based approaches under development.
Firstly, the use of a drug administered orally to patients, which has the ability to override a specific genetic mutation, called a nonsense mutation, has been used which introduces an abnormal stop signal into the CHM gene. Nonsense mutations account for over 30% of CHM.
Delivery of viral gene therapy to the retina of patients with CHM is currently in early phase 2 clinical trials in the UK, US and Canada to evaluate the efficacy of treatment.
Conclusion
Choroideremia is a rare genetic retinal disease, which causes blindness in males during middle age. We still have much to understand about how the biochemical defect causes the retinal-specific disease and if it has any hidden effects on the rest of the body.
Recently, there have been suggestions that CHM patients may be at risk of higher blood cholesterol or fat levels, and this is under currently investigation as part of detailed natural history studies.
CHM has been used as a prototype retinal degeneration for developing novel gene therapies and hence, has provided much hope for those affected with the disease but also patients with similar inherited retinal disorders.
Important links
For families, friends and patients with CHM, there are a number of organizations that can provide more information on the condition, research and support:
References
- Moosajee M et al. Clinical utility gene card for: choroideremia. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.183. PMID: 23963298
- Moosajee M et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum Mol Genet. 2016 Aug 15;25(16):3416-3431. PMID: 27329764 (Lay summary: http://atlasofscience.org/treating-nonsense-may-be-a-cure-for-the-blinding-disease-choroideremia/)
- MacLaren RE et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014 Mar 29;383(9923):1129-37. PMID: 24439297
Acknowledgements

Fight for sight - Someone in the world goes blind every five seconds. Our mission is to stop sight loss in its tracks. By funding pioneering eye research, we’re creating a future everyone can see.
- Moorfields Eye Hospital
- UCL Institute of Ophthalmology
- The Choroideremia Research Foundation
Further Reading
About Dr Mariya Moosajee
 Consultant Ophthalmologist, Moorfields Eye Hospital, Great Ormond Street Hospital for Children, and UCL Institute of Ophthalmology, London UK
Consultant Ophthalmologist, Moorfields Eye Hospital, Great Ormond Street Hospital for Children, and UCL Institute of Ophthalmology, London UK
Dr Mariya Moosajee is a Consultant Ophthalmologist specialising in Genetic Eye Disease at Moorfields Eye Hospital, Great Ormond Street Hospital for Children and UCL Institute of Ophthalmology.She graduated with First Class Honours in Biochemistry and Molecular Genetics in 2000, Medicine (MBBS) in 2003, and was awarded her PhD in Molecular Ophthalmology in 2009 all from Imperial College London.
Dr Moosajee is also a clinician scientist and leads her own research group at the UCL Institute of Ophthalmology. She is a Senior Clinical Lecturer and holds a Wellcome Trust Clinical Research Development Fellowship.
Her specialist interest is understanding the molecular basis of ocular maldevelopment and inherited retinal dystrophies, using zebrafish disease models and human induced pluripotent stem cell derived retinal cells. Through dissecting the molecular and cellular pathways of disease, Dr Moosajee and her team have identified potential therapeutic targets and developed treatment strategies, including nonsense suppression therapy, which is translational and applicable to a wide range of inherited eye disorders.
Dr Moosajee’s ultimate goal is to improve the lives of patients with genetic eye disease by directly improving genetic diagnosis, understanding the natural history of the disease using high resolution imaging techniques and developing novel treatments to counter these blinding diseases.
Disclaimer: This article has not been subjected to peer review and is presented as the personal views of a qualified expert in the subject in accordance with the general terms and condition of use of the News-Medical.Net website.
Last Updated: Jun 28, 2019
 World’s largest eye imaging and data resource expands across the UK
World’s largest eye imaging and data resource expands across the UK