SARS-CoV-2 led to self-limiting short infections with viral evolution over many consecutive rounds of infection, each interrupted by a transmission bottleneck. Superspreading events play a significant role in SARS-CoV-2 transmission. Such events involved conditions highly conducive to viral transmission and exhibited different patterns of viral evolution.
 Study: Narrow transmission bottlenecks and limited within-host viral diversity during a SARS-CoV-2 outbreak on a fishing boat. Image Credit: Natalia Dobryanskaya / Shutterstock
Study: Narrow transmission bottlenecks and limited within-host viral diversity during a SARS-CoV-2 outbreak on a fishing boat. Image Credit: Natalia Dobryanskaya / Shutterstock
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Study design
The current research work extracted ribonucleic acid (RNA) from a nasal swab of SARS-CoV-2-positive crew members, and primary sequencing libraries were constructed. With reverse transcription-quantitative polymerase chain reaction (RT-qPCR) cycle threshold (Ct) values less than 20, the researchers re-sequenced samples from original libraries by the amplification of viral RNA through PCR. The researchers obtained 1,113,690 average mapped reads from each library.
Data from raw sequencing files were handled by the Snakemake pipeline. SARS-CoV-2-specific reads were selected from the FASTQ files using 31 bases matching the Wuhan-1/2019 genomic reference utilizing Bestus Bioinformaticus Decontamination Using Kmers (BBDuk). Aligned binary alignment map (BAM) files were checked for quality using sequence alignment map (SAM) tools.
For sequencing analysis, the researchers assembled the consensus genome and arranged it with multiple alignments using Fast Fourier Transform (MAFFT). With an aligned genome, a phylogenetic IQ TREE with 1,000 bootstraps iterations was built. To include similar genomes that were present on the boat, a basic local alignment search tool (BLAST) database was constructed from all sequences in Washington during and before the outbreak.
Finally, the variants were identified using a custom Python Script. Single-nucleotide polymorphism (SNPs) were identified, and the total number of occurrences of SNPs were recorded and annotated for coding effect. The researchers also used three different variant calling programs - ivar, varscan2, and lofreq.
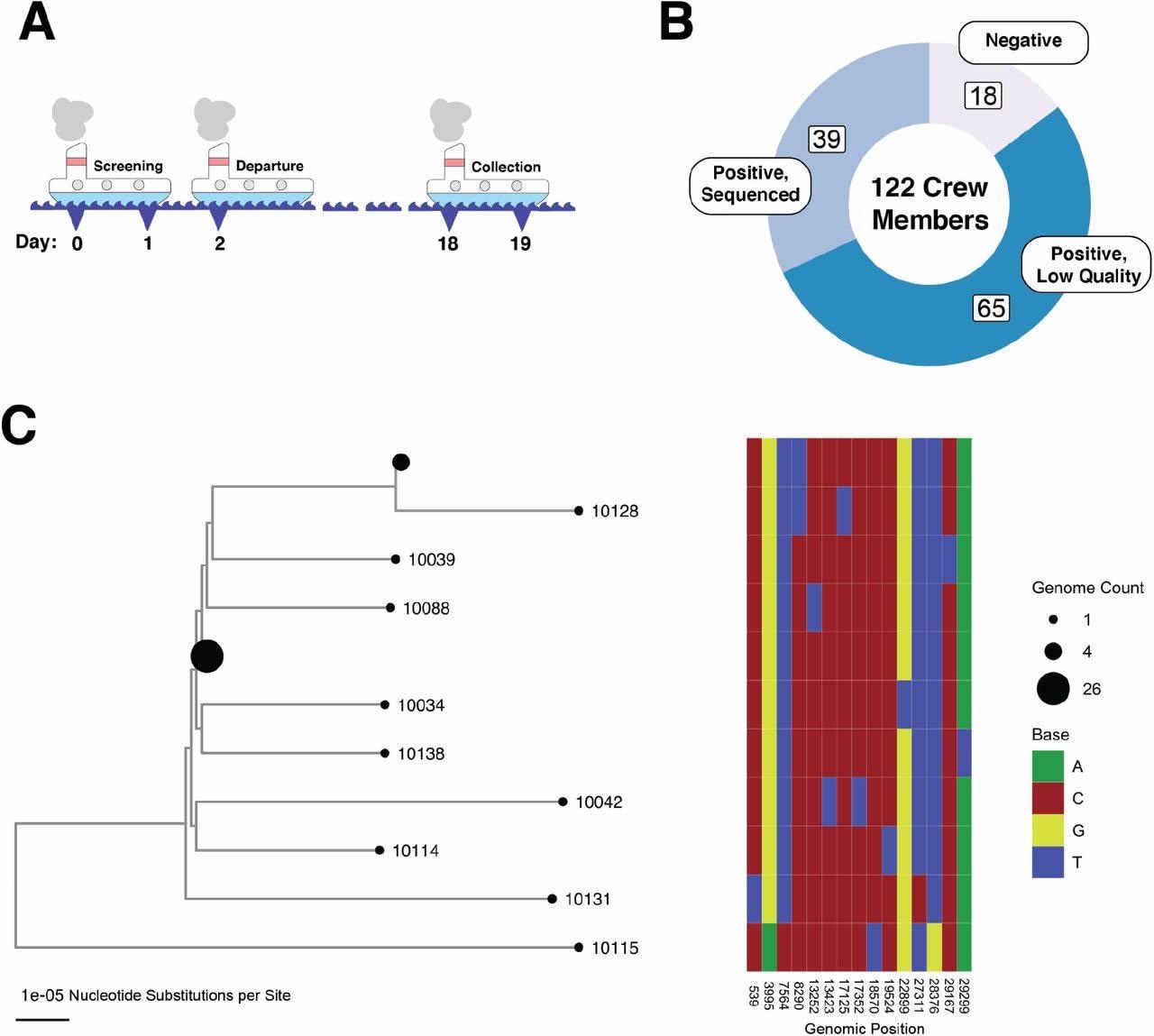
An outbreak of SARS-CoV-2 on an isolated fishing boat is an epidemiologically linked cluster of infections. (A) Schematic showing the timeline of the fishing vessel outbreak. All samples used in this study were taken on day 18 as shown in the figure (relative to the start of pre-departure screening). (B) Donut plot showing the sampling breakdown for all 122 members of the crew. (C) Phylogeny of SARS-CoV-2 genome from the boat. A heatmap to the right shows the nucleotide differences between genomes on the tree. Specimen identification numbers for crew member samples label the leaf nodes of the tree except for those nodes with more than one identical genome. Node sizes are proportional to number of sequences: there is a node representing 26 identical sequences (10101, 10126, 10133, 10105, 10108, 10130, 10031, 10110, 10030, 10124, 10029, 10102, 10038, 10094, 10027, 10118, 10117, 10106, 10091, 10093, 10127, 10116, 10040, 10090, 10036, 10089)and a node representing 4 identical sequences (10107, 10129, 10113, 10028); all other nodes represent unique sequences.
Findings
The results showed that during the pre-departure screening of 120 individuals from Seattle, none of them tested positive for SARS-CoV-2. However, after boarding the boat, 80% of crew members tested SARS-CoV-2-positive, indicating an elevation in a secondary attack rate. Interestingly, before the ship’s departure, neutralizing antibodies were present in only three crew members, and none of the individuals met the definition for infection.
Out of the samples of individuals who tested SARS-CoV-2-positive after returning the boat to shore, 39 nasal swab samples had sufficient high levels of viral RNA (Ct value < 26) used to assemble viral sequences from deep sequencing data. The team noted that 75% of viral sequences assembled from the boat were similar to at least one other sequence representing a superspreading event.
While performing high deep sequencing of samples, out of the 39 samples as above, 23 samples showed sufficient viral RNA (Ct value < 20) for metagenomic sequencing. The researchers used a stringent cut-off for depth sequencing with consideration of sequences having >80% of genome concealed by 100 reads in one or more replicates while performing downstream analysis. The team observed no biases across the viral genome length in sequence coverage and noted that the obtained results were robust for employing other methods for variant calling.
The researchers observed that within each crew member, the diversity of the virus population was limited, with a mean of three intra-host variants per member at a relatively low frequency, with only a few showing < 10 %. There was no correlation between nasal swab Ct values and the identified number of SNPs. Moreover, no discernable pattern was observed in the location of SNPs in the viral genome.
The researchers, while identifying the mutations, observed that the majority of low-frequency variants were specific or fixed to a single individual, and at intermediate frequencies, fixed variants were not observed. On the boat, four low-frequency alleles were identified in multiple individuals - A4229C, C9502T, G14335T, and T18402A - with not more than 5% frequency.
The variant C9502T was present in a stretch of thymine homopolymers, a known correlate of spurious variant calls in SARS-CoV-2 sequencing data. Moreover, G14335T and A4229C exhibited positional bias in the aligned reads. Finally, T18402A demonstrated significant frequency divergence between replicates. All of these characteristics of shared low-frequency variant alleles suggested that these alleles are technically sequencing artifacts rather than true mutations.
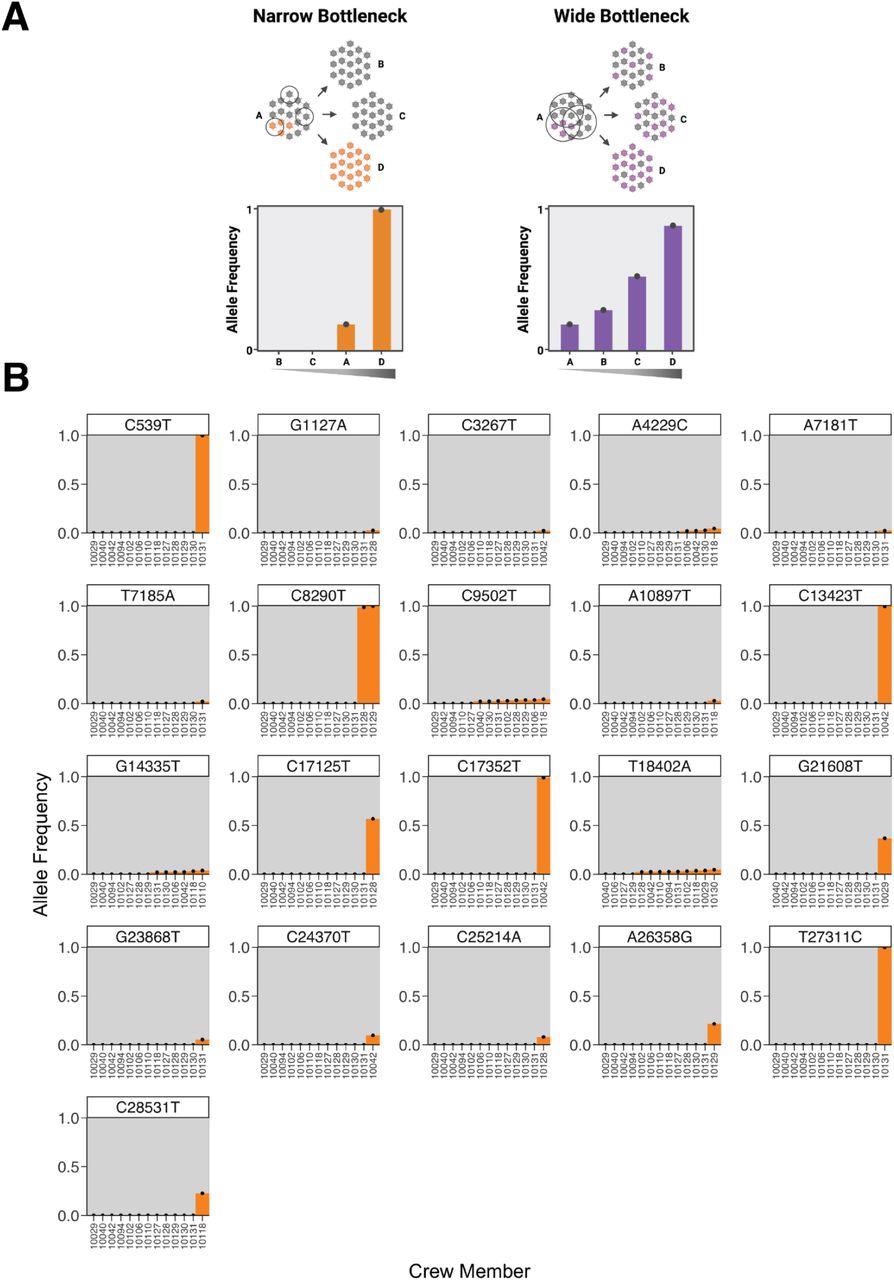
The spectrum of shared minor variation suggests that the transmission bottleneck is narrow. (A) A schematic showing the expected pattern of observed allele frequencies for shared variants in either a narrow or wide bottleneck scenario. (B) Each plot represents the frequency of a single nucleotide polymorphism (SNP) across crew members. Variants are called relative to the ancestral sequence of the virus introduced to the boat as inferred from the phylogeny of crew member genomes. The x-axis is ordered by variant frequency.
Conclusion
This research demonstrated that in a super spreading event of SARS-CoV-2 outbreak in a fishing boat, the epidemiologically linked individuals on the boat shared little or no intrahost viral diversity. Moreover, the evolution of the viral genome involved the fixation of mutations with low-frequency occasionally rather than the sustained maintenance of viral diversity within-host.
The observations of the current research work suggested that narrow transmission bottlenecks are a universal feature of the SARS-CoV-2 transmission. However, the present study has some limitations. For example, sequencing data were obtained only for a few crew members of the boat (13 out of 122), and there was a lack of quantitative estimation of the transmission bottleneck.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Narrow transmission bottlenecks and limited within-host viral diversity during a SARS-CoV-2 outbreak on a fishing boat. William W. Hannon, Pavitra Roychoudhury, Hong Xie, Lasata Shrestha, Amin Addetia, Keith R. Jerome, Alexander L. Greninger, Jesse D. Bloom. bioRxiv 2022.02.09.479546; https://doi.org/10.1101/2022.02.09.479546, https://www.biorxiv.org/content/10.1101/2022.02.09.479546v1
- Peer reviewed and published scientific report.
Hannon, William W, Pavitra Roychoudhury, Hong Xie, Lasata Shrestha, Amin Addetia, Keith R Jerome, Alexander L Greninger, and Jesse D Bloom. 2022. “Narrow Transmission Bottlenecks and Limited Within-Host Viral Diversity during a SARS-CoV-2 Outbreak on a Fishing Boat.” Virus Evolution 8 (2). https://doi.org/10.1093/ve/veac052. https://academic.oup.com/ve/article/8/2/veac052/6609212.
 Genetic discovery reveals biological cause of excessive sweating disorder
Genetic discovery reveals biological cause of excessive sweating disorder