A new study uncovers how diabetic red blood cells release toxic packages that damage blood vessels, revealing a promising path for preventing serious complications.
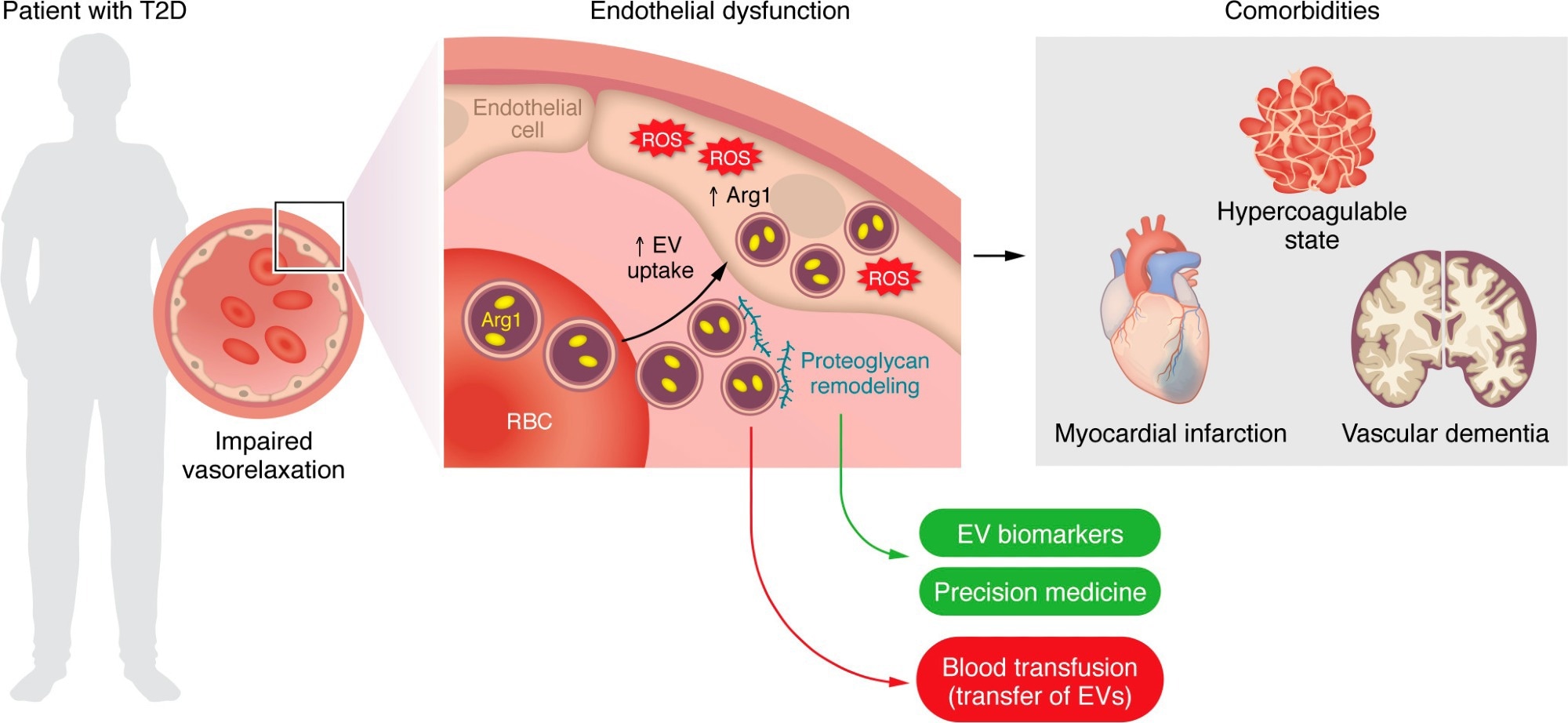 EVs derived from RBCs of patients with T2D are taken up by the endothelium and impair endothelium-dependent relaxation via an EV-mediated transfer of the prooxidant enzyme Arg1. Proteoglycan remodeling is a main event fostering EV uptake and may provide targets that could serve as disease biomarkers and/or personalized therapies for blocking EV uptake. RBC-EVs are also implicated in diabetes-related comorbidities, namely hypercoagulability, myocardial infarction, and vascular dementia. Blood transfusion from patients with cardiovascular risk factors represents a potential issue given the transfer of RBC-EVs and subsequent endothelial dysfunction in the recipient patient.
EVs derived from RBCs of patients with T2D are taken up by the endothelium and impair endothelium-dependent relaxation via an EV-mediated transfer of the prooxidant enzyme Arg1. Proteoglycan remodeling is a main event fostering EV uptake and may provide targets that could serve as disease biomarkers and/or personalized therapies for blocking EV uptake. RBC-EVs are also implicated in diabetes-related comorbidities, namely hypercoagulability, myocardial infarction, and vascular dementia. Blood transfusion from patients with cardiovascular risk factors represents a potential issue given the transfer of RBC-EVs and subsequent endothelial dysfunction in the recipient patient.
Type 2 diabetes mellitus (T2DM) is associated with many disabling and often fatal vascular complications, including stroke, heart attacks, limb gangrene, loss of sight, and kidney failure. Impaired endothelial function, caused by oxidative stress, underlies these events. The source of this stress has remained elusive.
A recent commentary published in the Journal of Clinical Investigation explains the importance of a study by Collado et al. (J Clin Invest. 2025;135(10):e180900) that demonstrates the central role of red blood cells (RBCs) in this phenomenon. This could promote the development of molecular therapies to prevent such complications.
Introduction
Until recently, most scientists thought that vascular dysfunction in T2DM was caused by endogenous endothelial factors. It now appears that it is caused by external factors.
One of these factors, a key contributor, is the RBCs. They enhance the level of the enzyme arginase-1 (Arg1), which promotes oxidation and elevates the levels of reactive oxygen species (ROS) within the endothelium. This, in turn, reduces the action of the vasodilator molecule, endothelial nitric oxide (NO).
T2DM RBC-EV release
In the referenced study, the scientists found that RBCs in T2DM patients secrete extracellular vesicles (EVs) carrying Arg1. These travel to and are taken up by the vascular endothelial cells. The resulting increase in endothelial Arg1 activity induces vascular damage with protean manifestations.
Diabetic RBCs release fewer EVs than healthy ones, yet only the former enter the endothelium. RBCs shed a fifth of their membrane area over their 120-day lifespan, so the mere release of RBC-EVs is not a marker of disease. The change in composition is more important, involving alterations to EV membrane heparan sulfate proteoglycans (e.g., syndecan-4, CD44).
These changes facilitate EV uptake. A better understanding of this could help develop treatments directed at the underlying mechanism of vascular disease in T2DM, such as inhibitors of proteoglycan remodeling. Such work could also help identify biomarkers of endothelial disease.
EV uptake and vasoconstriction
Endothelial uptake of RBC-EVs in T2DM patients results in impaired endothelium-dependent vascular relaxation compared with that from the RBCs of healthy patients.
Endothelial function improved when EV uptake was prevented by heparin, corroborating the role of EV uptake in endothelial dysfunction. Heparin, being a natural glycosaminoglycan, also confirms the importance of proteoglycan changes in EV uptake, especially as EVs express proteins including heparan sulfate proteoglycans.
Importantly, the study's proteomic analysis revealed other dysregulated ROS-related proteins in diabetic RBC-EVs, suggesting that Arg1 is part of a broader oxidative network.
Again, Arg1 inhibitor ABH prevented ROS increases after RBC exposure. Moreover, mouse aorta rings showed endothelial-dependent relaxation of the vessel wall despite being exposed to T2DM RBCs, in the presence of ABH. This provides evidence for the causal role of EV uptake in oxidative endothelial stress.
However, actual examination of the endothelium exposed to T2DM RBC-EV Arg1 in a living animal would better mirror actual tissue exposures in T2DM.
Genetic manipulations to silence or knock down endogenous Arg1 in endothelial cells confirmed that exogenous rather than endothelial Arg1 causes oxidative stress. This confirmed earlier work in which removing endothelial Arg1 in mice did not reverse endothelial dysfunction.
RBC-EVs from T2DM and healthy people have comparable Arg1 levels. Therefore, the increased EV uptake induces endothelial damage in T2DM, rather than EV release. How blood sugar levels affect endothelial EV uptake in T2DM is still unclear.
RBC-EVs and vascular disease
RBC-EVs also play a role in inducing hypercoagulability of blood, heart attacks, and vascular dementia in T2DM.
For instance, RBC-EVs in T2DM contain tissue factor, often alongside exposed phosphatidylserine residues, which promote clotting. RBC-EVs predict poorer outcomes in patients with ST-elevation myocardial infarction (STEMI) – the most common type of heart attack – after primary percutaneous coronary intervention (PCI), a procedure intended to restore blood flow.
Stored RBCs, particularly from diabetic donors, release EVs during storage, which could exacerbate risks when transfused into recipients.
Again, α-synuclein, a protein found in neuroinflammation and dementia, is elevated nearly 1,000-fold in RBC-EVs compared to cerebrospinal fluid. These EVs persist in the circulation and cross the blood-brain barrier, triggering brain inflammation. This may explain the injury to the capillaries and neurons of the brain in T2DM, causing the more rapid development of cognitive impairment and dementia.
Transfusing blood from such patients could potentially induce endothelial dysfunction and other adverse effects in the recipient. This deserves closer attention, especially when using blood from people older than 65 years or smokers, who have elevated cardiovascular risk.
Conclusions
“Evidence provided by this study lets us believe that the solution to the problem could not be in the vascular endothelium, but in the blood.” The pathbreaking study both points out the mechanism of endothelial dysfunction in T2DM and suggests novel molecular targets to prevent its occurrence.
Journal references:
- Commentary: Costantino, S., Mohammed, S. A., and Paneni, F. (2025). Endothelial dysfunction in patients with type 2 diabetes: the truth is in the blood. Journal of Clinical Investigation. doi: 10.1172/JCI193128, https://www.jci.org/articles/view/193128
- Primary study: Collado A, et al. (2025). Erythrocyte-derived extracellular vesicles induce endothelial dysfunction through arginase-1 and oxidative stress in type 2 diabetes. J Clin Invest. 135(10):e180900, DOI: 10.1172/JCI180900, https://www.jci.org/articles/view/180900