Modeling indicates that secondary structures of RNA in the genes encoding the Nsp4 and Nsp16 proteins of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are different from other related coronavirus species, and this may affect some viral molecular processes.
With the dramatic spread of COVID-19 caused by the SARS-CoV-2 coronavirus, there has been a push to understand how a virus can infect new hosts and what makes it different from other coronaviruses.
One way of doing this is to figure out which parts of the viral genome have been naturally selected to evolve and are different from ancestor species and which parts have been selectively removed from the genome.
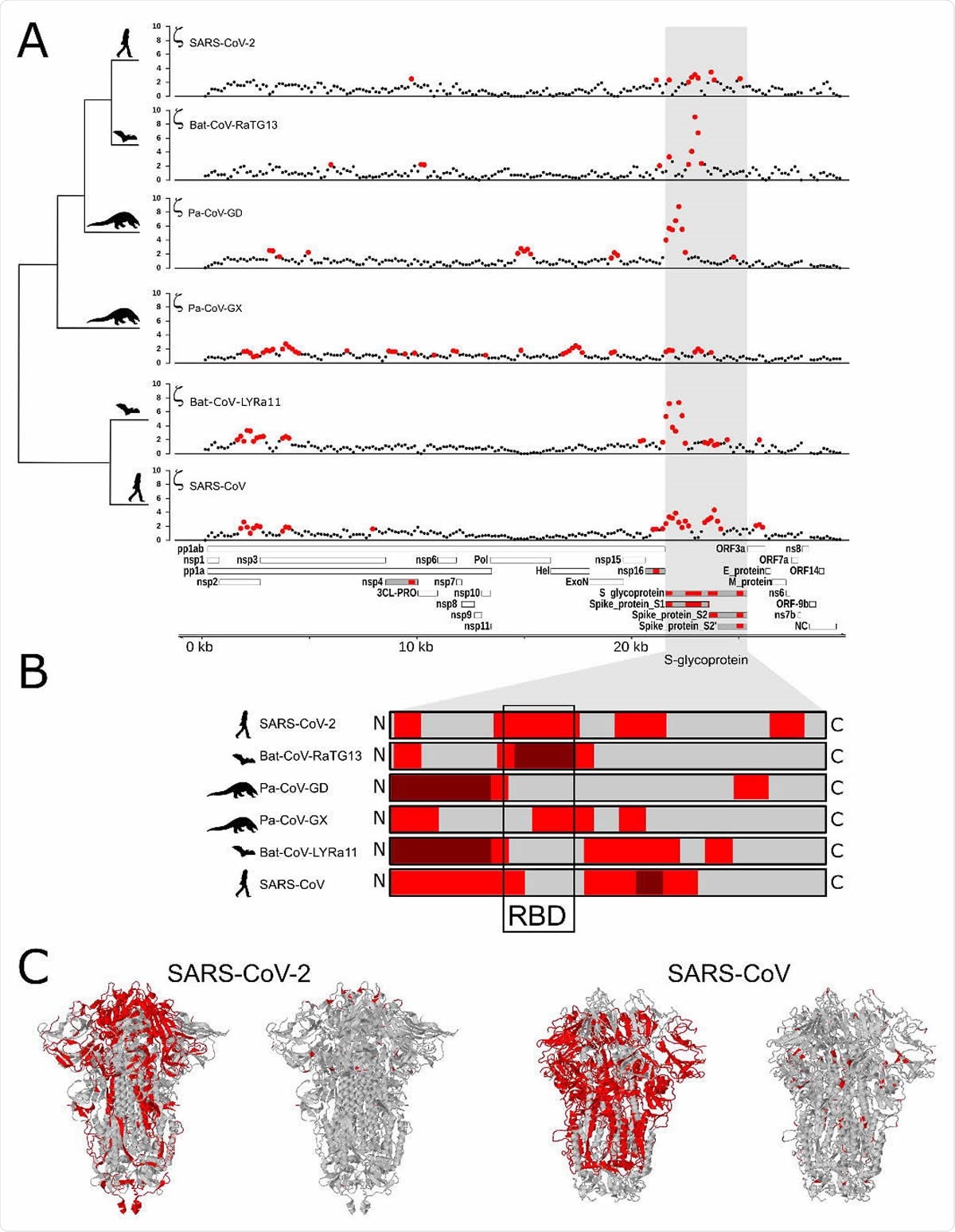
Previous studies have found a mixture of selective evolution and removal in the genes that encode the spike protein of the SARS-CoV-2 coronavirus. The spike protein helps the virus invade and infect the host cell by attaching to the angiotensin-converting enzyme 2 (ACE2).
However, there are other critical processes in RNA viruses, like the coronavirus, that are not controlled by the protein sequences. Standard tests to determine mutations can detect changes in the viral proteins, but do not include RNA molecules that the proteins interact with for different processes.
Mutations found in spike protein
To investigate mutations in coronaviruses, a new study by scientists at Duke University and published on the preprint server bioRxiv*, used a computational methodology, adaptiPhy, which identifies extra nucleotide substitutions in certain parts of the viral genome compared to mutations that have no effect on the genome. Using adaptiPhy, the team identified regions of genomes of different Sarbecovirus species from bat, pangolin, and human hosts, which could have been positively selected, or advantageous mutations. For the novel SARS-CoV-2 coronavirus, they used about 5000 genome sequences from the NCBI Virus database.
The team also investigated changes in the Nsp4 and Nsp16 protein structures at the RNA and protein level using modeling.
Using different computational methods, the researchers found the most prominent signal was for the gene that encodes the spike protein, showing positive selection in all the species tested. This is similar to what other studies have reported.
In the SARS-CoV-2 virus, they found positive selection in four regions of the spike protein gene. One was a change in the entire receptor-binding domain (RBD) structure, which binds to host cells. Another was a change in the site that is necessary for infecting lung cells.
This is different from the changes found in SARS-CoV and Bat-CoV-LYRa11. In these viruses, positive selection occurred in the regions for viral camouflage and those that allow virus entry into the host cell.
These mutations suggest that the viruses adapted for different hosts, with the SARS-CoV-2 adapting to bind to the ACE2 protein in various hosts.
Positive selection in proteins
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
The authors also found positive selection in the genes encoding two proteins, Nsp4 and Nsp16, something not seen before. In Nsp4, they found two nucleotide substitutions, valine to alanine and valine to isoleucine. Modeling suggested these changes did not have any significant impact on the secondary or tertiary structure of the protein in SARS-CoV-2 compared to other species. In Nsp16, they did not find any such substitutions. However, none of these changes likely affect the structure or functions in these proteins, write the authors.
So, it's possible the positive selection was due to changes in the structure and function of RNA.
Nsp16 has a single well-folded region, which is the only such region that is also conserved in other related coronavirus species. Nsp4 has two somewhat well-folded regions. Thus, it's likely that these folded structures are related to viral functions.
"Our minimum free energy (MFE) predictions reveal that the likely secondary structure of the RNA genome in the region of the Nsp4 and Nsp16 genes likely differs among the six coronavirus species we examined," write the authors.
There were also differences among species in the entropy in the regions of positive selection, suggesting differences in the stability of the folded molecules.
"Together, these new results indicate that the folded regions of Nsp4 and Nsp16 in the SARS-Cov-2 genome may differ in shape from those of related coronaviruses," the authors write.
However, how these changes, which are unique to SARS-CoV-2, are linked to specific molecular functions, cannot be determined yet, as the molecular functions of secondary structures in coronaviruses are not well known today.
Since previous studies indicate these regions have functional roles, the changes may affect genome or transcript functions. However, the true roles of these adaptations in the structural proteins need to be experimentally studied further.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Berrio A. et al. (2020) Positive selection within the genomes of SARS-CoV-2 and other Coronaviruses independent of impact on protein function. bioRvix. https://doi.org/10.1101/2020.09.16.300038
- Peer reviewed and published scientific report.
Berrio, Alejandro, Valerie Gartner, and Gregory A. Wray. 2020. “Positive Selection within the Genomes of SARS-CoV-2 and Other Coronaviruses Independent of Impact on Protein Function.” PeerJ 8 (October): e10234. https://doi.org/10.7717/peerj.10234. https://peerj.com/articles/10234/.
 SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity
SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity