Over 2.2 million genomic sequences of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have been generated since the start of the coronavirus disease 2019 (COVID-19) pandemic.
As of June 5, 2021, these sequences have been and deposited and shared internationally through the online database GISAID. The analysis of these sequences provides an insight into the evolution and epidemiology of the virus.
 Study: Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. Image Credit: ktsdesign / Shutterstock.com
Study: Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. Image Credit: ktsdesign / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
The Pango nomenclature system
A total of four different nomenclature systems have been used by scientists to name and identify genetically distinct SARS-CoV-2 types and linages. Originally developed in early 2020, the Pango dynamic nomenclature system has since become a widely used tool for SARS-CoV-2 classification.
Pango, which is short for Phylogenetic Assignment of Named Global Outbreak Lineages, examines the phylogenetic structure of the ongoing pandemic at high resolution. The Pango nomenclature is designed to classify complete or near-complete SARS-CoV-2 genomes. Taken together, the Pango system contains over 1,300 lineages that cover the entire genetic diversity of SARS-CoV-2.
However, an incomplete SARS-CoV-2 genome sequence that consists of the complete nucleotide sequence of the spike protein, is generated. The spike protein contains the virus’ receptor-binding domain (RBD), which binds to the human host cell receptor angiotensin-converting enzyme 2 (ACE2) with high affinity. As a result, the RBD of the S protein is the primary target of natural and vaccine-elicited immunity.
These incomplete sequences, which are otherwise known as spike-only nucleotide sequences (SNSs), raise important questions on how Pango reliably designates the genome to a lineage and assigns the spike-only nucleotide sequences. In a recent study published on the preprint server bioRxiv*, a group of researchers determined the degree to which Pango lineages are distinguishable using spike-only nucleotide sequences, with a focus on current SARS-CoV-2 variants of concern (VOCs).
About the study
Mutations of the spike protein may be unique or shared among different SARS-CoV-2 lineages. The researchers explored how these spike mutations observed at differing frequencies are used in the classification of spike-only genomes.
Because the SNSs are shared among tens or hundreds of Pango lineages, the researchers introduced the notion of designating such sequences to a “lineage set” to represent the range of Pango lineages that are consistent with mutations in a given SNS. This can support the development of software tools that can assign newly-generated SNSs to Pango lineage sets.
Study findings
In the current study, the researchers extracted SNSs from the full SARS-CoV-2 genomes with Pango designations. The genome sequences of the SARS-CoV-2 variants were then compared to the original SARS-CoV-2 strain in an effort to identify all consensus spike haplotypes (CSH) and lineage sets.
Among the SARS-CoV-2 genomes that were identified, the researchers found that over 89% of the amino acid positions in the spike protein varied between these genomes. While there are three possible genetic variations that can arise within the sike protein, the researchers found that 34% were synonymous genetic variations, whereas non-synonymous changes and indel changes were both considerably rare. Taken together, these observations indicate the SNSs may contain enough information that can be used to distinguish a SARS-CoV-2 strain from other Pango lineages.
The analysis of each of the Pango lineages led to the observation that a small number of spike mutations were observed in all, or nearly all, of these sequences. Additionally, distinct SNSs have been found in high numbers in each of the Pango lineages, with the same SNS often identified in multiple Pango lineages. There is also a high variance in the number of SNSs that were identified in each Pango lineage.
The analysis of SNSs in the Pango lineages ultimately led to the conclusion that are insufficient as a method to classify each Pango lineage. As a result, the researchers of the current study turned to CSHs.
Although a majority of CSHs will be unique to one Pango lineage, others will be shared between multiple lineages. As a result, the researchers created the concept of a “lineage set,” which describes a set of lineages that share the same CSH.
Each lineage set must be designed using CSHs that are defined with a mutation frequency threshold (X) that is lower than 100%. Ultimately, this approach is easier to curate and interpret as compared to when SNSs alone are used.
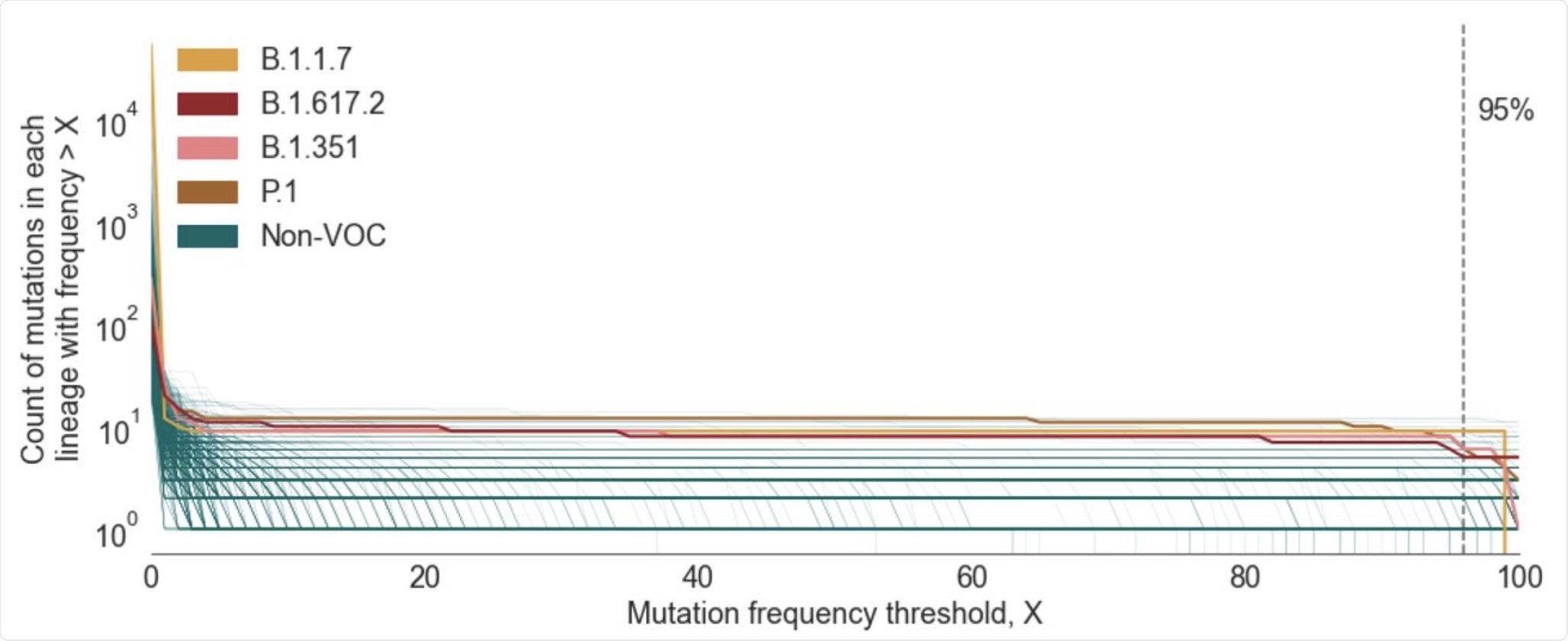 Plot showing the number of mutations in the CSH for a given lineage as a function of the mutation frequency threshold (X) used to define the CSH. Lineages shown are those that contain >20 designated sequences with complete spike nucleotide sequences and >5 spike mutations. The lineages that correspond to the four main VOCs are colored individually, whilst all other included lineages are shown in green.
Plot showing the number of mutations in the CSH for a given lineage as a function of the mutation frequency threshold (X) used to define the CSH. Lineages shown are those that contain >20 designated sequences with complete spike nucleotide sequences and >5 spike mutations. The lineages that correspond to the four main VOCs are colored individually, whilst all other included lineages are shown in green.
Conclusions
The researchers wrote a python-based script that can output lineage set assignments based on a custom pangoLEARN model trained on spike sequences. This platform eliminates any potential conflicting information that would otherwise arise when only SNSs are utilized. This tool is currently free and available to the public at https://github.com/aineniamh/hedgehog.
“We, therefore, developed a system by which spike-only sequences can be designated to a “lineage set” that contains all of the Pango lineages consistent with the CSH) of that sequence.”
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
O’Toole, A., Pybus, O. G., Abram, M. E., et al. (2021). Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. bioRxiv. doi:10.1101/2021.08.10.455799. https://www.biorxiv.org/content/10.1101/2021.08.10.455799v1.full.
- Peer reviewed and published scientific report.
O’Toole, Áine, Oliver G. Pybus, Michael E. Abram, Elizabeth J. Kelly, and Andrew Rambaut. 2022. “Pango Lineage Designation and Assignment Using SARS-CoV-2 Spike Gene Nucleotide Sequences.” BMC Genomics 23 (1): 121. https://doi.org/10.1186/s12864-022-08358-2. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08358-2.
 Severe COVID-19 reactivates dormant viruses and may leave a lasting viral signature
Severe COVID-19 reactivates dormant viruses and may leave a lasting viral signature