In a recent study published in the United States Centers for Disease Control and Prevention (CDC) Emerging Infectious Diseases journal, researchers investigate the spread and transmission ability of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lineages B.1.619 and B.1.620 in South Korea.
Background
Over two million genomes of SARS-CoV-2 have been identified since the beginning of the coronavirus disease 2019 (COVID-19) pandemic, thus demonstrating that this virus is highly mutational.
Studies have shown that the SARS-CoV-2 spike protein attaches itself to the host cell angiotensin-converting enzyme 2 (ACE2) receptor during infection. Mutations in the spike protein receptor-binding domain (RBD) have led to the emergence of several SARS-CoV-2 variants of concern (VOC).
SARS-CoV-2 VOCs identified by the World Health Organization (WHO) are known to cause serious health implications. These variants included Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529). Each of these variants exhibits higher transmissibility, evades the immune system, is more likely to cause severe symptoms, reduce the effectiveness of vaccines, and escape detection.
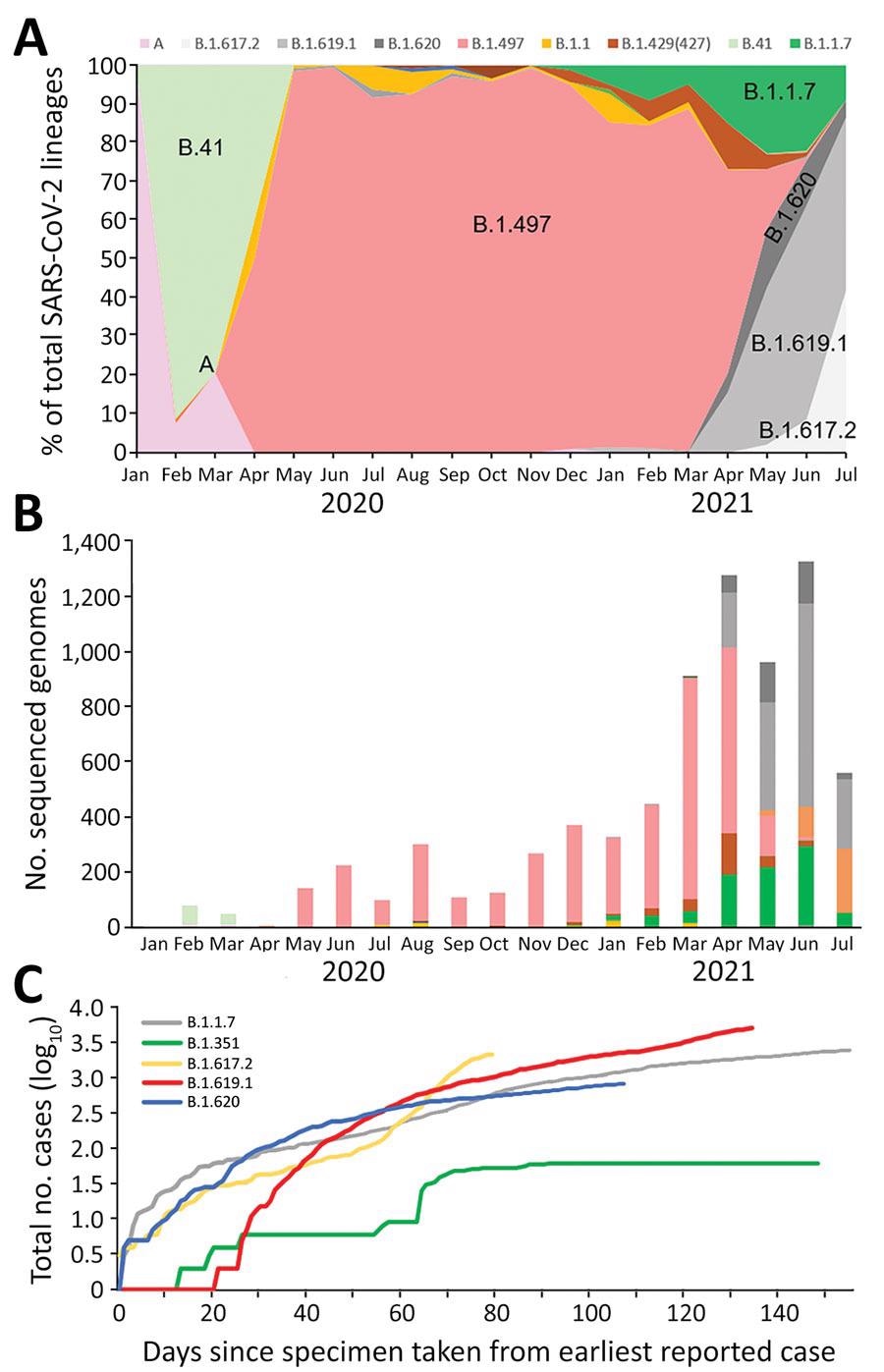
Investigation of SARS-CoV-2 B.1.619 and B.1.620 lineages, South Korea, 2021. A) Distribution of 7 SARS-CoV-2 lineages in domestic cases over time. Data are shown for lineages A, B.41, B.1.497, B.1.1.7 (Alpha variant), B.1.617.2 (Delta variant), B.1.619.1, and B.1.620. B) Number of sequenced genomes over time, by lineage. C) Logarithmic graph of cumulative cases of variants indexed by days since the first reported case as of July 21, 2021. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
About the study
In the current study, researchers investigate the constant mutation of SARS-CoV-2 and the emergence of new variants to efficiently monitor the effect of variants on disease severity and other factors affecting public health.
The researchers collected nasopharyngeal and oropharyngeal swab samples from COVID-19-positive patients who were tested using real-time reverse transcription PCR (rRT-PCR). These samples were used to prepare whole-genome sequencing (WGS) libraries and were later sequenced appropriately. The researchers then aligned whole genomic sequences for phylogenetic tree analysis, where the maximum-likelihood phylogenetic trees were inferred and visualized.
The researchers acquired SARS-CoV-2 WGS libraries of more than 5% of the total reported COVID-19-positive cases as of July 21, 2021. They filtered out total positive cases from the period of April to July 2021, which is when the number of individuals infected with SARS-CoV-2 lineages B.1.619 and B.1.620 exhibited a sharp increase.
A and B.41 lineages were most commonly found at the beginning of the pandemic in South Korea, while the B.1.497 lineage became prevalent in March 2020. In June 2021, the prevalence of the Alpha variant increased to 22%, whereas the B.1.619 and B.1.620 variants reached 11.5%, and the B.1.497 lineage decreased from 94.3% to 0.9%.
Study findings
The researchers found that the B.1.619 and B.1.620 lineages show several distinctive spike protein mutations but share the E484K mutation. These specific mutations in the spike protein have been found to significantly influence SARS-CoV-2 pathogenesis.
Other than the E484K mutation, the B.1.619 lineage has N440K mutations, which might make the lineage resistant to monoclonal antibodies and enhance its binding affinity to the ACE2 receptor. The B.1.620 lineage has S447N substitutions, which help evade antibody-mediated immunity while also improving RBD affinity for ACE2.
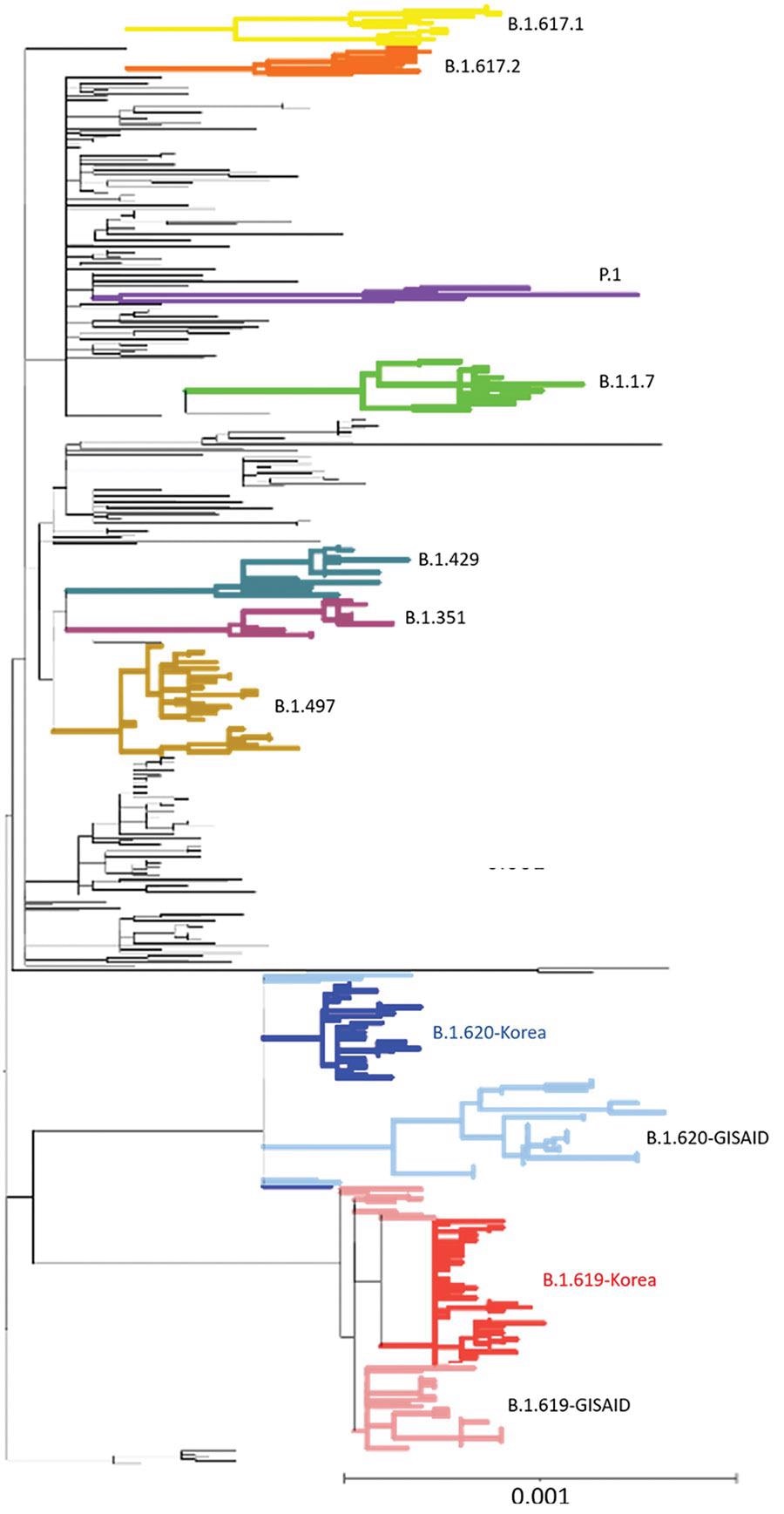
Phylogenetic analysis of severe acute respiratory syndrome coronavirus 2 sequences, South Korea. A total of 457 sequences were used to construct the tree, including 37 sequences of B.1.619 lineage and 36 sequences of B.1.620 lineage from GISAID (https://www.gisaid.org). Each sequence was aligned to the reference sequence (Wuhan-Hu-1, GenBank accession no. NC_045512) using Geneious Prime software (https://www.geneious.com) and then manually trimmed to equal lengths. A maximum-likelihood phylogenetic tree was reconstructed using FastTree version 2.1.9 (http://www.microbesonline.org/fasttree), under the general time-reversible plus gamma nucleotide substitution model; the phylogenetic tree was visualized using iTOL (https://itol.embl.de). Four variants of concern (B.1.1.7, B.1.351, P.1, and B.1.617.2), 2 variants of interest (B.1.429 and B.1.617.1), and B.1.497, which were the major lineages of the GH clade in South Korea, are shown. Red indicates South Korea B.1.619 sequences and blue, B.1.620; pink indicates Europe B.1.619 sequences and light blue, B.1.620. Scale bar indicates substitutions per site.
The authors also noted that the B.1.619 and B.1.620 lineages do not inhibit the neutralizing activity in vaccinated or convalescent individuals. The combined effect of the lineages on factors like viral pathogenicity and transmissibility needs to be determined in further investigations.
Conclusion
The current study findings demonstrate the various mutations and transmissibility of different SARS-CoV-2 variants in South Korea. Importantly, the transmissibility of the B.1.619 and B.1.620 lineages, as well as their effect on disease severity, is not thoroughly investigated in the study.
According to the researchers, the neutralizing capacity of people who had recovered from non-VOC infections and vaccinated individuals is sufficient to combat infection by these lineages.
 Scientists identify common symptom patterns in post-COVID-19 vaccination syndrome
Scientists identify common symptom patterns in post-COVID-19 vaccination syndrome