As of March 7, 2022, the ongoing coronavirus disease 2019 (COVID-19) pandemic, which is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused over six million deaths worldwide.
Respiratory viral infection rates are often exacerbated by sleep and circadian rhythm dispersion (SRCD). In particular, shift workers are more likely to encounter SRCD, which predisposes them to an increased risk for respiratory viral infections. Studies on SARS-CoV-2 have provided evidence of the links between shift work causing SRCD and the risk of contracting COVID-19.

Study: Sleep and circadian rhythm disruption alters the lung transcriptome to predispose to viral infection. Image Credit: LightFiield Studios / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
A recent study published in the bioRxiv* preprint server explores the effect of SRCD on mouse lung transcriptome after six of hours sleep deprivation.
Study findings
The findings depicted an enhancement in circadian rhythm genes, steroid hormone feedback, kinase activity, and cellular processes facilitating viral entry and ribonucleic acid (RNA) replication.
On analyzing the sleep-deprived (SD) downregulated genes, repression of immune system pathways was observed. In particular, SD was found to upregulate furin, which modulates the entry of SARS-CoV-2 by cleaving the spike protein, while it downregulates Toll-like receptors (TLRs), which trigger innate immune responses that can also affect the pathogenesis of COVID-19.
An escalation in the proinflammatory transcription factor Nfkbia, which is a major down-regulator of nuclear factor-kappa (NF-kB) by 46% and its repressor Tle1 by 43% were found after sleep deprivation. Twelve genes were found to upregulate SD, whereas 23 genes were downregulated by SD, the latter of which positively affects NF-kB function.
Amongst the SD downregulated genes, 215 gene ontology biological pathways (GOBP) terms were significant, of which 72% were immune system pathways, 23% were adaptive immunity specific terms, and 18% were innate immune specific terms. These findings are suggestive of a suppressed immune system impacting pathways that are involved in viral entry, replication, and trafficking due to sleep deprivation.
Out of 2,029 cycling genes, 911 were deranged by SD. Analysis of 3,532 non-rhythmic-SD differential genes revealed that many were directly affected by SD but were not circadian regulated.
Comparatively, an analysis of the rhythmic-SD differential genes reflected an increased circadian regulated gene expression. Dysregulation of rhythmic pathways like signaling, metabolism, protein folding, post-translational protein modification, and RNA processing was observed after SD, all of which contribute to disrupted circadian lung physiology.
An attempt to discern the magnitude of circadian rhythm disruption revealed an alteration in many circadian transcripts following SD, thereby suggesting a compromise in the time-dependent genetic expression and coherent core molecular circadian rhythm. Analysis of the principal component of lung transcriptomes revealed disrupted circadian networks following SD. The results demonstrate an alteration in circadian regulation following SD, which increases susceptibility to viral infections in the respiratory tract.
How these results compare to previous findings
The researchers also cross-referenced their results with three independent studies. Out of the 50 genes investigated by Daniloski et al., ten were dysregulated post SD, out of which eight were found to participate in SARS-CoV-2 replication. Out of the 32 genes involved in the viral entry as identified by Zhu et al., eight were found to be SD differential genes and consisted of four upregulated and four downregulated genes.
A comparison of the 50 host factors mentioned by Wei et al. to SD differential genes of the study revealed an intersection of 11 genes, eight of which were upregulated genes primarily involved in transcriptional regulation. These observations demonstrate amplification of processes and host factors after acute SD that affect the SARS-CoV-2 life cycle.
Out of the 332 human-virus protein-protein identified by Gordon et al., 87 overlapped with SD differential genes of the study, 40 of which were recognized to function in viral replication, 18 affected SARS-CoV-2 immune evasion, with several others impacting SARS-CoV-2 infection. These findings suggest a differential expression of many host factors post SD that may potentiate SARS-CoV-2 replication.
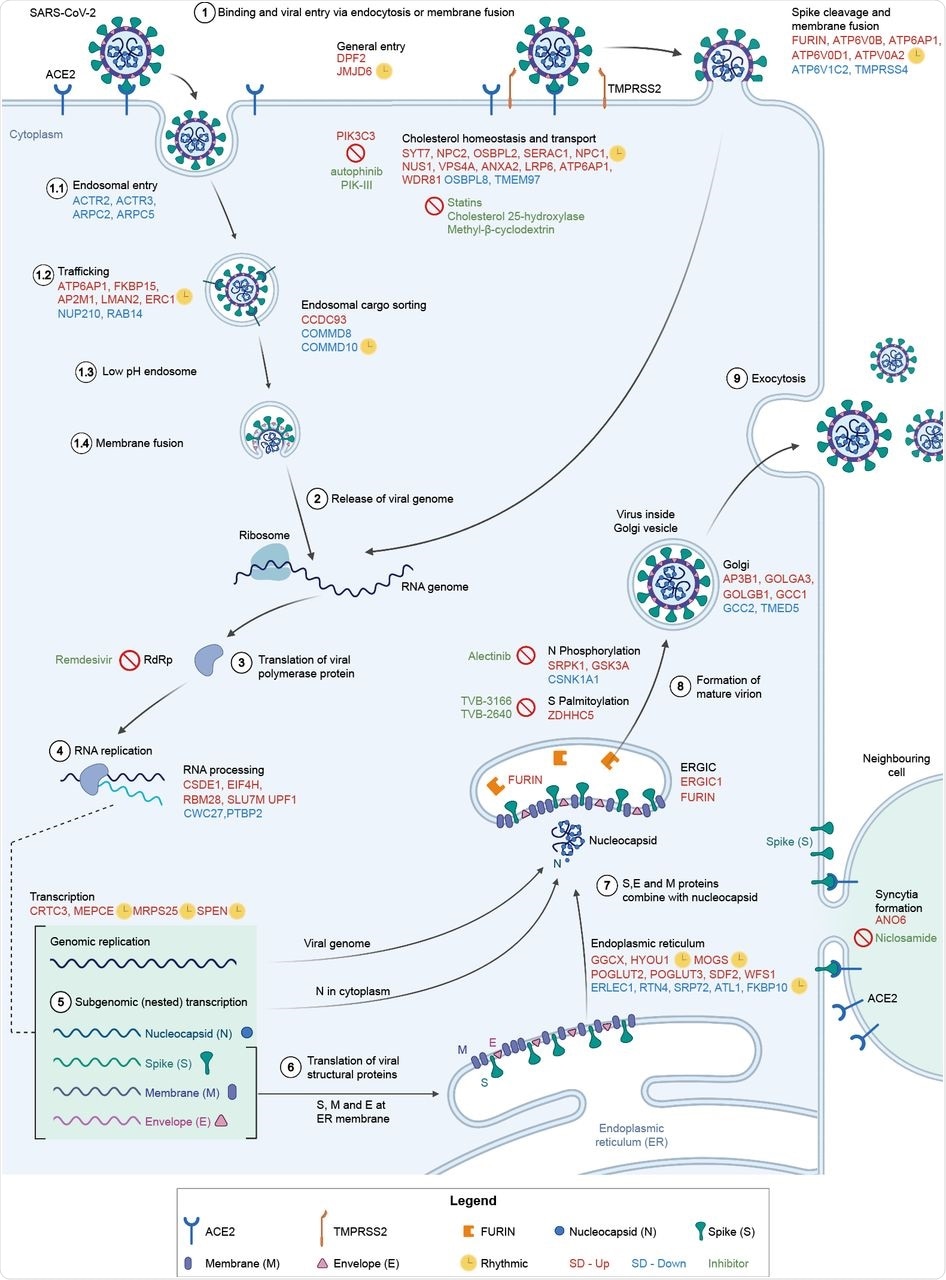
Involvement of differentially expressed genes after sleep deprivation in the SARS-CoV-2 life cycle. (1) SARS-CoV-2 binds ACE2 and enters via endocytosis or membrane fusion, depending on the availability of TMPRSS2/4. (2) The viral RNA genome is released into the cytoplasm and (3-4) replicated and (5) transcribed by RdRp. (6) Viral structural proteins are translated by host ribosomes. (7-8) The virion assembles and (9) is released. All differentially expressed genes shown (red font for SD-Up, blue font for SD-Down) apart from FURIN, TMPRSS4, GSK3A, SRPK1, and CSNK1A1 are critical host factors overlapping with at least one of the studies from Gordon et al. (2020b), Daniloski et al. (2021), Wei et al. (2021), or Zhu et al. (2021). Drugs targeting SD differential or viral genes mentioned are in green font. Cycling genes are denoted by a yellow clock. ACE2 = angiotensin-converting enzyme 2, ERGIC = ER-Golgi apparatus intermediate compartment, RdRp = RNA-dependent RNA polymerase, TMPRSS2 = transmembrane protease serine 2. Adapted from Du et al. (2009) and from “Coronavirus Replication Cycle” by BioRender.com. Created with BioRender.com.
Implications
The current study proposed several mechanistic pathways, following which SD-differential genes facilitate entry, replication, and trafficking of SARS-CoV-2. SD dysregulated all pathways involving extracellular transmembrane protease serine-4 (Tmprss4), Furin, Atp6v0d1, Atp6ap1, and Atp6v0b. Other pathways including endosome recycling, transcriptional modulation, and endolysosomal fusion were also dysregulated post SD.
Overall, 13 genes encoding intracellular cholesterol trafficking were differentially expressed post SD. Decreased morbidity and mortality were associated with statins, which could also reduce recovery time. However, the mechanism by which cholesterol affects SARS-CoV-2 pathogenesis remains unclear.
SD affected post-translational protein modification regulating SARS-CoV-2 replication. For example, the genes encoding phosphorylation of viral nucleocapsid were differentially expressed after SD.
A reduction in Zdhhc5 led to depalmitolylation of spike proteins compromising the membrane fusion, as well as viral entry; however, SD was found to increase the transcription of Zdhhc5. These findings suggest an increase in SARS-CoV-2 replication post SD.
The data also revealed suppression of immune-associated genes after SD that allow for viral persistence. For example, SD was found to alter the expression of RNF41 and TBKBP1, which regulate interferon production, are targeted by SARS-CoV-2 proteins.
SARS-CoV-2 was also found to use the NK-kB pathway as an immune evasion strategy by causing differential expression of Tle1, Trim59, and E3 ubiquitin ligases Mib1. SARS-CoV-2 evades the innate immune response by exploiting the ubiquitination machinery. In fact, six SD differential genes that were implicated in ubiquitination code for proteins, have been found to interact with proteins of SARS-CoV-2.
Of note, Ano6, which regulates syncytia formation in the lungs, appeared to be upregulated following SD. Five SD-impacted mitochondrial host genes engaging SARS-CoV-2 protein-protein interactions were also identified. Taken together, these findings highlight immune evasion mechanisms by SARS-CoV-2 after acute SD.
Conclusions
The current study confirmed alterations in mice lung transcriptomes following sleep deprivation, which could explain the increased risk of severe SARS-CoV-2 infection and other respiratory viral infections after SCRD, which is often associated with shift work. Taken together, these findings could guide therapeutic approaches that could prevent and treat SARS-CoV-2 infection.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Taylor, L., Von Lendenfeld, F., Ashton, A., et al. (2022). Sleep and circadian rhythm disruption alters the lung transcriptome to predispose to viral infection. bioRxiv. doi:10.1101/2022.02.28.482377. https://www.biorxiv.org/content/10.1101/2022.02.28.482377v1.
- Peer reviewed and published scientific report.
Taylor, Lewis, Felix Von Lendenfeld, Anna Ashton, Harshmeena Sanghani, Simona Di Pretoro, Laura Usselmann, Maria Veretennikova, et al. 2023. “Sleep and Circadian Rhythm Disruption Alters the Lung Transcriptome to Predispose to Viral Infection.” IScience 26 (2): 105877. https://doi.org/10.1016/j.isci.2022.105877. https://www.cell.com/iscience/fulltext/S2589-0042(22)02150-2.
 Study maps how exercise, sleep, diet, and mood connect in daily life
Study maps how exercise, sleep, diet, and mood connect in daily life