A recent study has revealed a full cycle of substrate cleavage for the main protease of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) – providing, in turn, a steadfast foundation for developing potent new viral inhibitors. The paper was published in the journal Proceedings of the National Academy of Sciences (PNAS).
Although vaccines were indispensable in our battle against coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2, new drugs are needed in order to lessen the burden of disease even more.
Thus far, substantial efforts have been concentrated on targeting the evolutionarily conserved nonstructural proteins of SARS-CoV-2, which play a role in viral replication and transcription – two important mechanisms of viral propagation.
Among them, main protease (usually abbreviated as Mpro) is one of the most attractive therapeutic targets due to its decisive role in replicase polyprotein cleavage and the fact that it is evolutionarily conserved across all coronaviruses.
One salient step toward the rational design of selective and potent inhibitors that target Mpro can be accomplished by discerning detailed mechanisms underlying the process of proteolysis (i.e., protein breakdown) and substrate recognition.
In this new study, a research group led by Dr. Yao Zhao from the ShanghaiTech University in China successfully determined the structure of viral protease in the resting state but also in the pre-cleavage state and post-cleavage state during protein breakdown.
A multipronged methodological approach
In order to fully grasp the substrate preference of Mpro in processing polyproteins, this research group initially determined the structure of Mpro mutant strain of SARS-CoV-2 (H41A) in complex with a series of substrates derived from multiple cleavage sites on polyproteins.
An extensive cloning process, protein expression, and purification of SARS-CoV-2 ensued, and the researchers have measured the binding affinity between the aforementioned H41A mutant and the six substrates.
Furthermore, crystallization, data collection and structure determination were done with specific methods and software support. Finally, microscale thermophoresis (MST) assays were carried out with the fluorescent labeling of the H41A mutant.
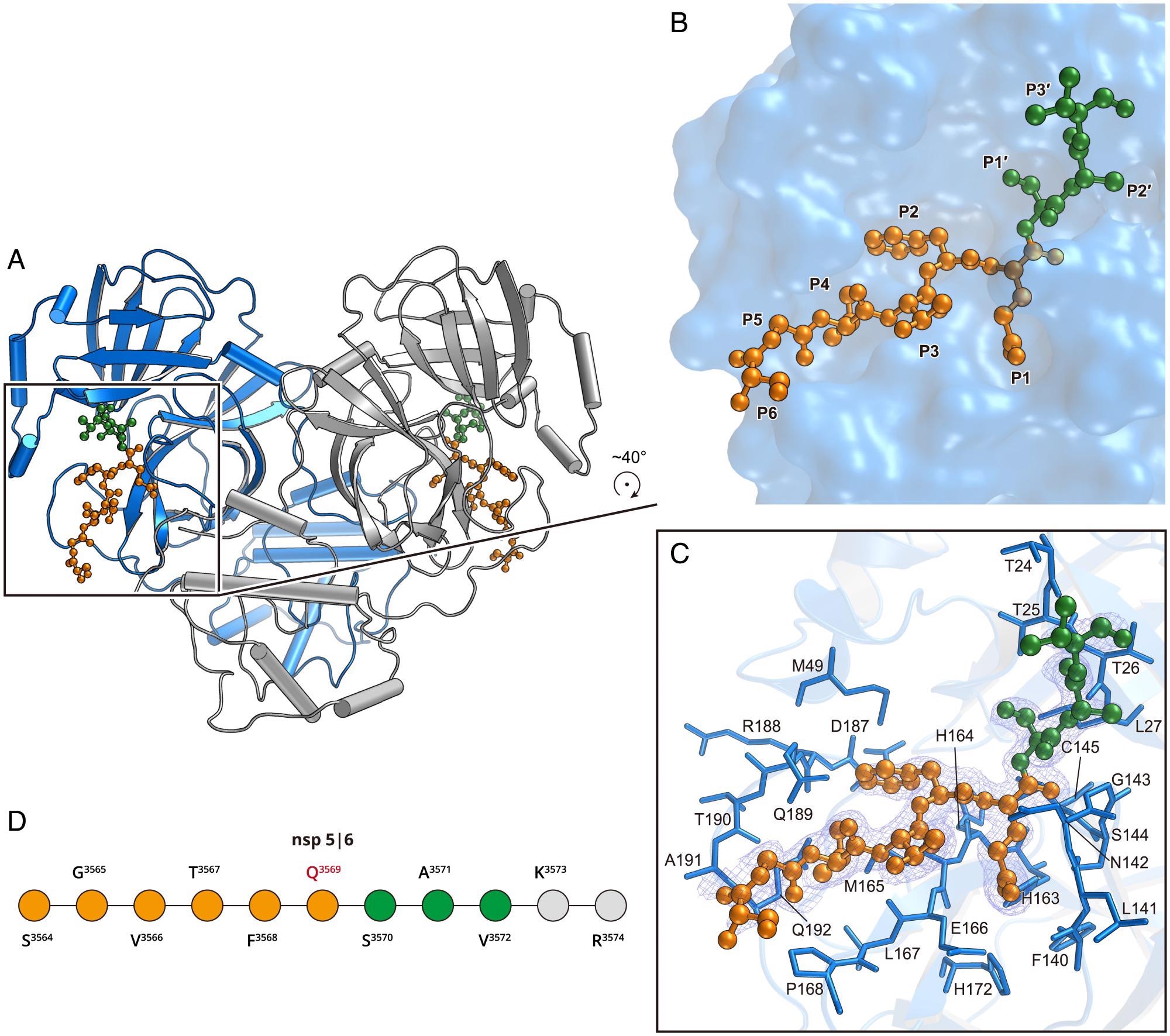 Structure of the H41A mutant in complex with the nsp5|6 peptidyl substrate. (A) The overall structure of H41A–nsp5|6 in dimer form. Protomer A and protomer B are colored blue and gray, respectively. (B) The zoom-in view of the substrate-binding pocket. The nsp5|6 peptidyl substrate is shown as a ball-and-stick model. Residues from P1 to P6 and P1′ to P3′ are colored in orange and green, respectively. (C) The detailed interaction between Mpro and its cleavage substrate. Residues involved in the substrate binding are shown as marine sticks. The polder map colored as blue mesh is contoured at 2.5σ. (D) The schematic diagram of nsp5|6. Residues that can be traced according to the electron density map are colored in orange or green. Residues that cannot be traced are in gray.
Structure of the H41A mutant in complex with the nsp5|6 peptidyl substrate. (A) The overall structure of H41A–nsp5|6 in dimer form. Protomer A and protomer B are colored blue and gray, respectively. (B) The zoom-in view of the substrate-binding pocket. The nsp5|6 peptidyl substrate is shown as a ball-and-stick model. Residues from P1 to P6 and P1′ to P3′ are colored in orange and green, respectively. (C) The detailed interaction between Mpro and its cleavage substrate. Residues involved in the substrate binding are shown as marine sticks. The polder map colored as blue mesh is contoured at 2.5σ. (D) The schematic diagram of nsp5|6. Residues that can be traced according to the electron density map are colored in orange or green. Residues that cannot be traced are in gray.
Flexibility in substrate recognition
In short, this study has shown that SARS-CoV-2 Mpro can recognize substrates as long as ten residues, with the special selectivity for four subsites (S1, S1′, S2, and S4). Alongside the most conserved S1 subsite, a very adaptable S2 subsite is also crucial for drug design efforts.
The findings have further shown that domain III of Mpro represents the most flexible domain, which can adjust its conformation to allow its C terminus to access the substrate binding pockets from its surrounding active membrane proteins.
The flexibility of this region is quite important for regulating its C-terminal autocleavage process (which means cleavage without the use of enzymes). More specifically, the Mpro C terminus in orientation 2 may enable favorable insertion into the substrate-binding pocket of its neighbor Mpro during C-terminal proteolysis.
Finally, this suggests that small molecules which can block the conformational change of domain III may actually be used as allosteric inhibitors for Mpro – a potentially very important addition to our armamentarium against COVID-19.
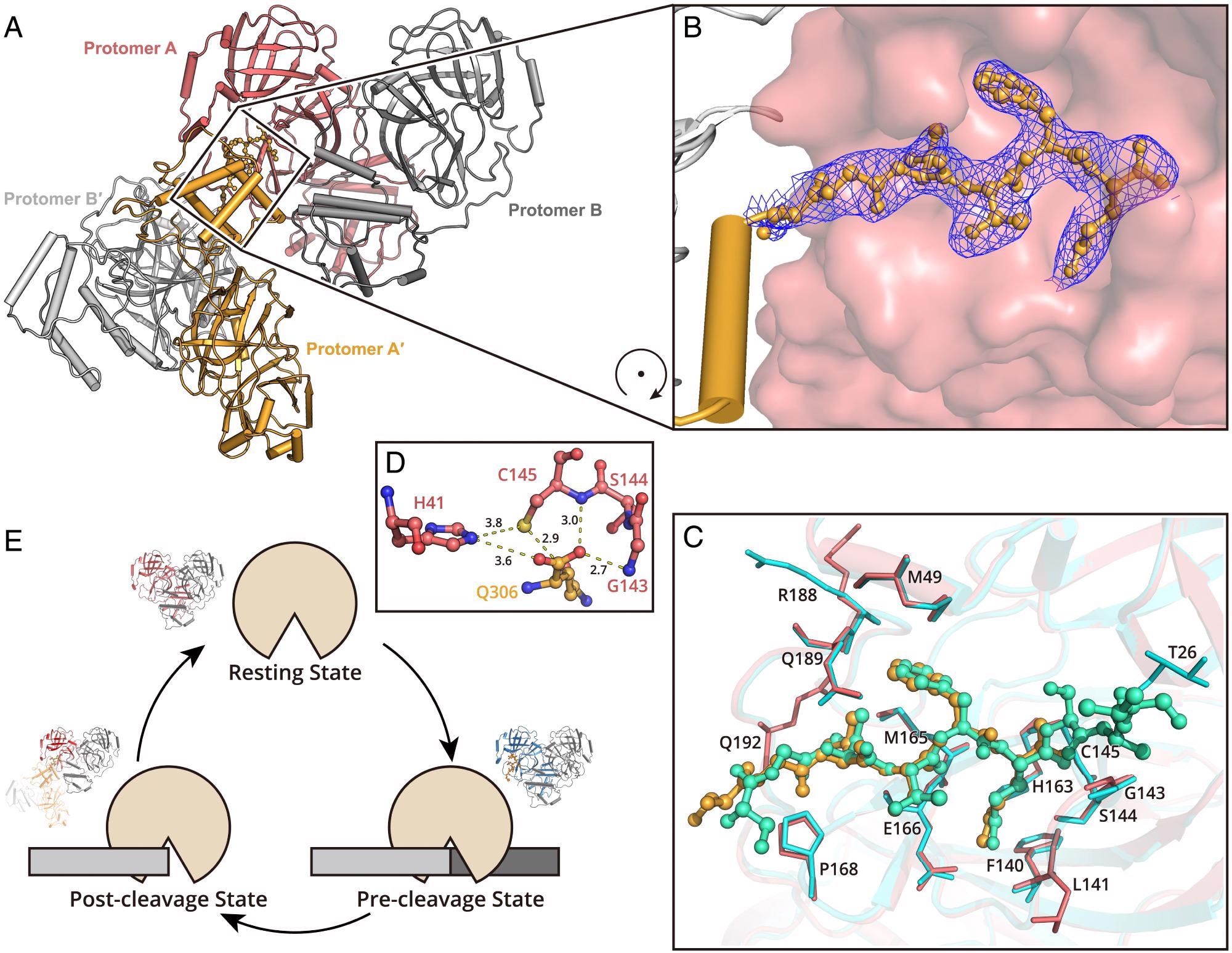 Structure of SARS-CoV-2 Mpro in the postcleavage state. (A) Two Mpro dimers are associated together, representing the postcleavage state. One dimer pair is colored in salmon (protomer A) and gray (protomer B). A second dimer is formed by protomer A′ and B′ (bright orange and light gray). The C terminus of protomer A′ is inserted into the substrate binding pocket of protomer A. (B) The zoom-in view of the C terminus of protomer A′. The residues S301 to Q306 are shown as a ball-and-stick model. The 2Fo–Fc density map contoured at 1.0σ is shown in blue mesh. (C) A comparison of the H41A–nsp5|6 complex structure and Mpro in postcleavage state structure. The H41A–nsp5|6 complex is colored in cyan and green (protease in cyan and peptidyl substrate nsp5|6 in green), and Mpro in postcleavage state is colored in salmon and bright orange (protease in salmon and C-terminal protomer A′ in bright orange). Substrates located in substrate binding picket are shown as ball-and-stick models. Residues involved in substrate binding are shown as sticks. (D) The arrangement of amino acids in the catalytic dyad. (E) A cartoon visualization of the cleavage cycle of SARS-CoV-2 Mpro.
Structure of SARS-CoV-2 Mpro in the postcleavage state. (A) Two Mpro dimers are associated together, representing the postcleavage state. One dimer pair is colored in salmon (protomer A) and gray (protomer B). A second dimer is formed by protomer A′ and B′ (bright orange and light gray). The C terminus of protomer A′ is inserted into the substrate binding pocket of protomer A. (B) The zoom-in view of the C terminus of protomer A′. The residues S301 to Q306 are shown as a ball-and-stick model. The 2Fo–Fc density map contoured at 1.0σ is shown in blue mesh. (C) A comparison of the H41A–nsp5|6 complex structure and Mpro in postcleavage state structure. The H41A–nsp5|6 complex is colored in cyan and green (protease in cyan and peptidyl substrate nsp5|6 in green), and Mpro in postcleavage state is colored in salmon and bright orange (protease in salmon and C-terminal protomer A′ in bright orange). Substrates located in substrate binding picket are shown as ball-and-stick models. Residues involved in substrate binding are shown as sticks. (D) The arrangement of amino acids in the catalytic dyad. (E) A cartoon visualization of the cleavage cycle of SARS-CoV-2 Mpro.
Implications for drug development
Even though many designed or screened inhibitors that are able to target the substrate-binding pocket of Mpro have been either developed or discovered, rather potent inhibitors with clinical potential are still necessary.
“Highly selective inhibitors against Mpro could be potential broad-spectrum drug candidates to fight against coronavirus infections not only for today but also in the future,” say study authors in this PNAS publication. “Structural data presented here provide a basis to develop potent new inhibitors against SARS-CoV-2”, they add.
In any case, more work will be needed if we are to translate these findings from bench to bedside, but high-resolution insights in the structural data are quite encouraging and definitely a step in the right direction.
 COVID-19 virus not retained in placenta after maternal recovery
COVID-19 virus not retained in placenta after maternal recovery