Various studies have reported novel SARS-CoV-2 mutations that have been associated with increased transmissibility, increased binding to angiotensin-converting enzyme 2 (ACE2), and antibody evasion. However, the functional consequences of such mutations and their links to the fitness of SARS-CoV-2 are still unknown.
 Study: Inferring selection effects in SARS-CoV-2 with Bayesian Viral Allele Selection. Image Credit: NIAID
Study: Inferring selection effects in SARS-CoV-2 with Bayesian Viral Allele Selection. Image Credit: NIAID
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
About the study
In the present study, researchers developed Bayesian viral allele selection to determine the genetic factors that influence differential viral fitness as well as the growth rates of different SARS-CoV-2 variants.
The Bayesian viral allele selection (BVAS) developed by the team allowed the computation of the posterior inclusion probability (PIP). It was noted that alleles having high PIPs were good candidates for influencing viral fitness. The team conducted comparisons between three methods based on viral diffusion, including mean apparent propagator (MAP), BVAS, and Laplace.
The team assessed the sensitivity of BVAS to hyperparameters such as the prior inclusion probability h and the prior precision τ. The value of PIP with respect to BVAS was also demonstrated by examining the allele-level sensitivity and precision observed when the alleles having PIPs more than 0.1 were considered as hits. Furthermore, the team estimated the relative viral fitness of all SARS-CoV-2 variants by fitting the BVAS model to allele frequencies found in different regions.
Results
The study results showed that in the analysis of the four viral diffusion methods, the causal hit rate increased with the number of regions and decreased as the number of alleles increased. The BVAS methods displayed the best hit rates among the four methods, while the efficiency of the MAP and Laplace methods was significantly low in the presence of a high number of alleles.
The sensitivity of BVAS to τ was slightly more than four orders of magnitude; however, the sensitivity reduced when the value of τ was very high. The team also observed that the value of effective population size (ν) played a crucial role in the BVAS sensitivity. Large values of ν indicated that increments in allele frequency were dependent on the deterministic drift. On the other hand, small values of v suggested that allele frequency increments displayed significant variability that was predominant in the deterministic drift. When the team considered alleles with PIPs over 0.1 as hits, high precision was observed for BVAS. This indicated that alleles with high PIP values were more likely to be causally associated with viral fitness. Moreover, the effective population size was found to decline by 15 folds as the sampling rate (ρ) reduced from 64% to 1%.
Estimating the fitness of SARS-CoV-2 lineages showed that SARS-CoV-2 Omicron BA.2 was the fittest lineage, followed by Omicron BA.1, Delta, Alpha, and the wild-type variant. Notably, some of the phylogenetic assignment of named global outbreak (PANGO) lineages exhibited diverse genotypes that corresponded to distinct growth rates. The team also remarked that the Omicron variant had fractured into various sublineages with fitness levels that have improved over time. Omicron BA.2.12.2 sublineage was found to be the fittest lineage, while other BA.2 sublineages also have comparable fitness levels.
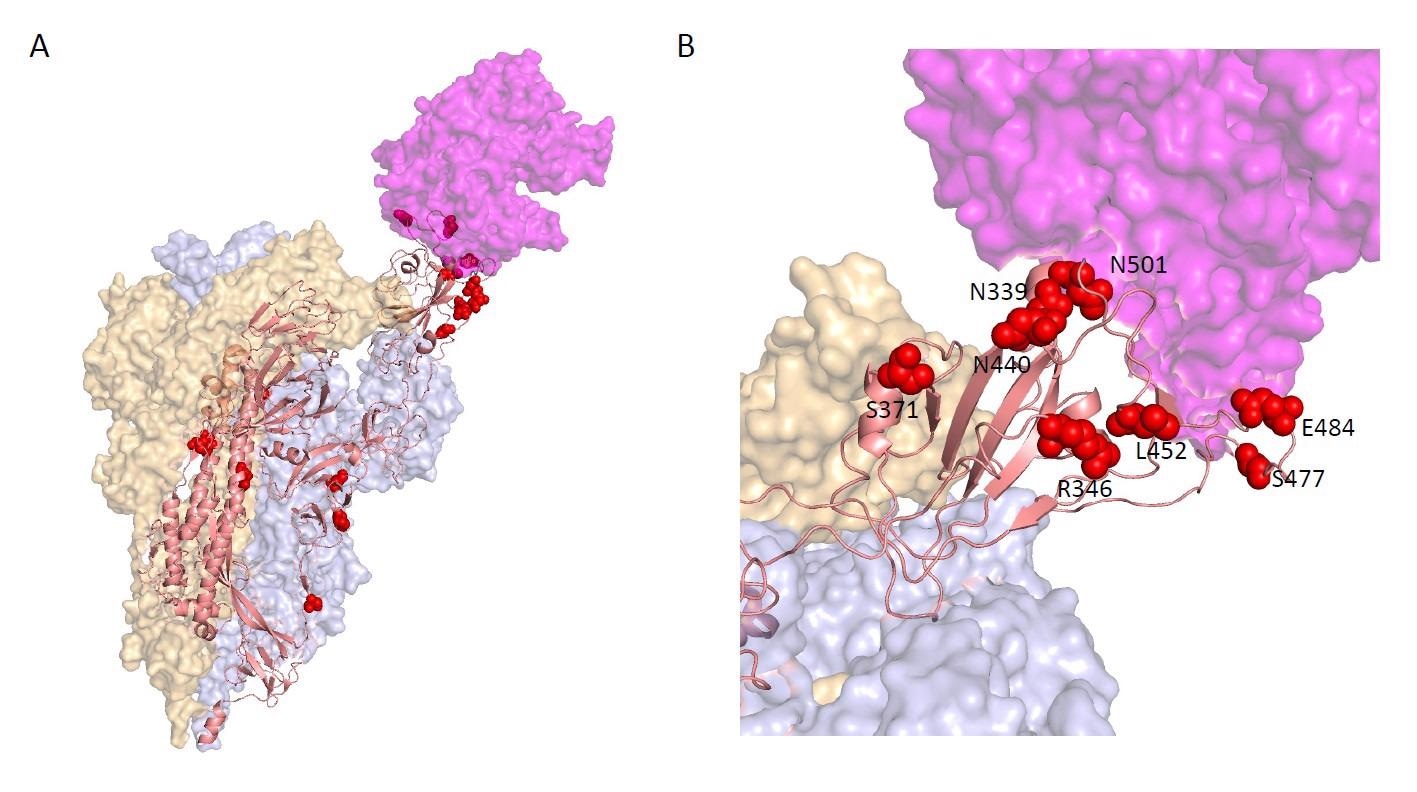 Locations of the top 20 Spike hits, ranked by PIP, on the Cryo-EM structure of a Spike trimer bound to ACE2 (magenta) at 3.9 Angstrom resolution in the single RBD "up" conformation from (Zhou et al., 2020) B. Enlarged view of the RBD-ACE2 interface, showing the spatial proximity of S:R346, S:N339, S:N440, S:L452, S:S477, S:E484, and S:N501.
Locations of the top 20 Spike hits, ranked by PIP, on the Cryo-EM structure of a Spike trimer bound to ACE2 (magenta) at 3.9 Angstrom resolution in the single RBD "up" conformation from (Zhou et al., 2020) B. Enlarged view of the RBD-ACE2 interface, showing the spatial proximity of S:R346, S:N339, S:N440, S:L452, S:S477, S:E484, and S:N501.
The team also found recombinant lineages, which were a result of recombinations between BA.1 and BA.2, and Delta and BA.1. Among these, XN and XT were the fittest recombinants; however, their fitness level was lower than that of BA.2 and higher than that of BA.1. Moreover, the fitness of existing recombinants such as XA-XT indicated that the fitness of these recombinant lineages might not be a subject of concern in the near future.
The analysis of SARS-CoV-2 mutations showed that the most robust selection signal was in the spike (S) protein, with the highest concentration of signals in the receptor-binding domain (RBD). Strong selection signals were also detected in the N-terminal domain (NTD) as well as the furin cleavage sites. Taking the effect size into account, the S:L452R mutation was found to be the highest hit and was found in the lineages BA.4/BA.5, B.1.427 and B.1.429. Also, the S:L452Q mutation had one of the highest hits and was found in the BA.2.12.2 variant.
Overall, the study findings showed the importance of the Bayesian viral allele selection method in understanding the selection effects of SARS-CoV-2 and its emerging variants.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
 SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity
SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity