The atmosphere is the most untouched microbial habitat on the Earth, and airborne bacteria are the most complex and dynamic communities influencing the Earth’s microbiomes. There are more than 1 × 104 bacterial cells/m3 and hundreds of unique taxa in the air. Large-scale studies have systemically documented the microbial features in soil, ocean, and human waste. Also, they have suggested an interrelationship between airborne microbiomes and surface environments. However, there is a lack of studies documenting airborne microorganisms, especially concerning their community structure.
Microbes do not live in isolation. Instead, they have multiple ecological relationships, ranging from mutualism to competition. Thus, determining their biogeographic distribution patterns and interactions with Earth’s other microbiomes, which define their origins, could shed light on the effects of changing climate/environment and anthropogenic activities.
About the study
In the present study, researchers first developed a global airborne bacterial dataset to assess their degree of commonality and interrelationships. This dataset comprised 76 newly gathered air particulate samples combined with 294 samples collected for previous studies from 63 sites worldwide. The sampling sites were diverse in terms of altitude and geography and encompassed ground level to rooftops (from 1.5m to 25 m high) to mountains 5,380 m above sea level, densely populated urban cities, and the remote Arctic Circle.
The team obtained the dataset for comparison from the Earth Microbiome Project (EMP), which accumulated 5,000+ samples from 23 surface environments. The airborne bacterial reference catalog had more than 27 million non-redundant 16S ribosomal RNA (rRNA) gene sequences.
Further, the researchers constructed a global airborne community co-occurrence network encompassing 5,038 significant correlation relationships (Spearman’s ρ > 0.6) among 482 connected operational taxonomic units (OTUs). OTUs are analytical units grouped by DNA sequence similarity in microbial ecology. Finally, the team used structural equation modeling (SEM) to explore the mechanisms driving microbial communities. Likewise, they calculated the total effects of environmental filtering and bacterial interactions on shaping communities.
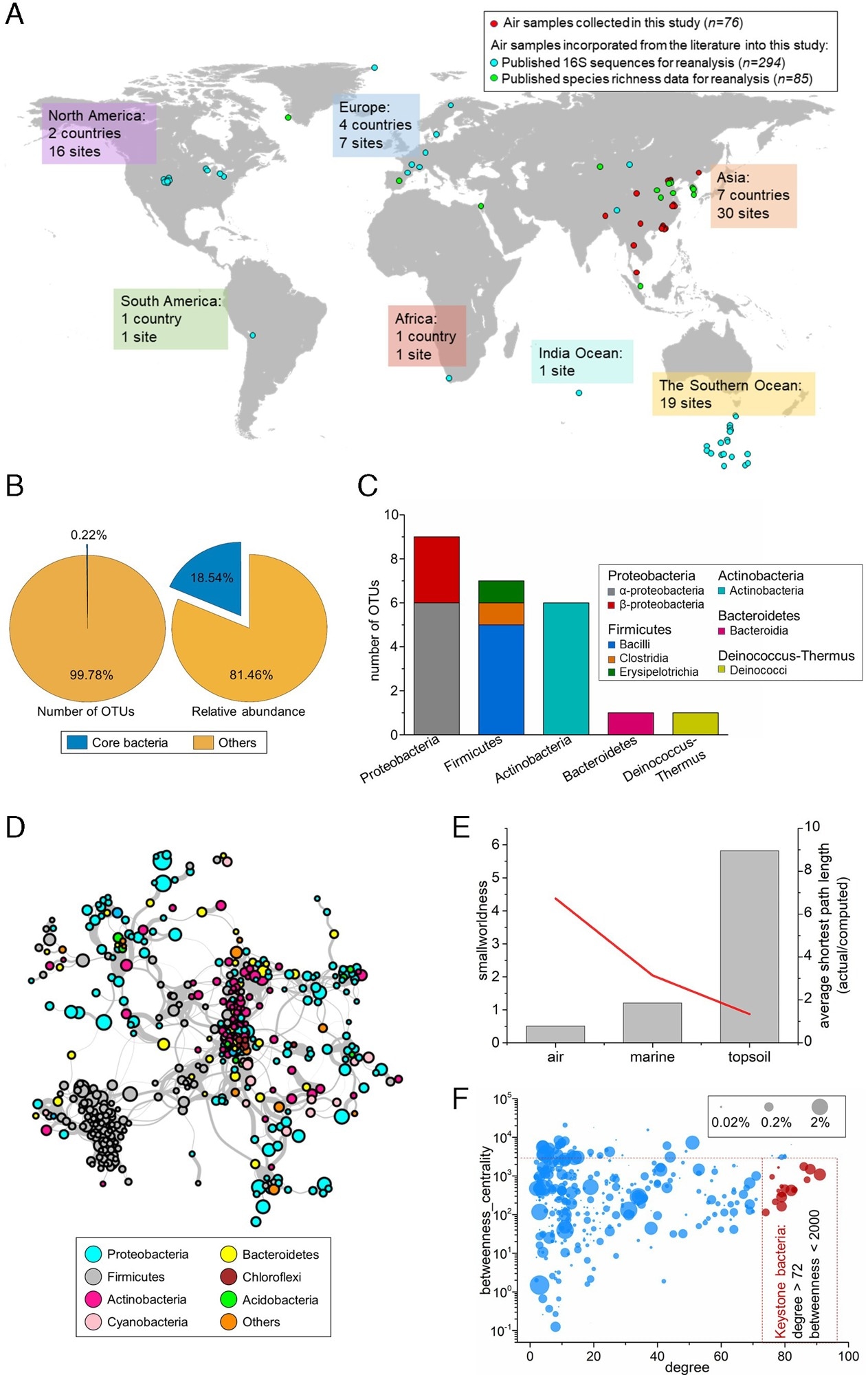 The structure of globally distributed airborne bacterial communities. (A) Locations where air samples and environmental data were collected across the globe. (B) The number, proportion, and relative abundance of the global core OTUs compared with those of the remaining bacterial OTUs. (C) The taxonomic composition of the global core bacteria at the phylum and class level. (D) The global airborne bacterial community co-occurrence network. The connections (edges) stand for a strong (Spearman’s ρ > 0.6) and significant (p < 0.01) correlation. The nodes represent the combined OTUs with unique annotations for genus level in the datasets. The size of each node was proportional to the mean relative abundance across 370 samples. Nodes were colored by the phyla of the bacteria. (E) “Small-network” identification based on a “smallworldness” index and the average shortest path length of the global bacterial community network in the air, marine, and soil environments. (F) Degree—the betweenness centrality plot of each node in the co-occurrence network. The nodes in red are viewed as keystone species. The size of the nodes shows the relative proportions of the OTUs in the total microbiome.
The structure of globally distributed airborne bacterial communities. (A) Locations where air samples and environmental data were collected across the globe. (B) The number, proportion, and relative abundance of the global core OTUs compared with those of the remaining bacterial OTUs. (C) The taxonomic composition of the global core bacteria at the phylum and class level. (D) The global airborne bacterial community co-occurrence network. The connections (edges) stand for a strong (Spearman’s ρ > 0.6) and significant (p < 0.01) correlation. The nodes represent the combined OTUs with unique annotations for genus level in the datasets. The size of each node was proportional to the mean relative abundance across 370 samples. Nodes were colored by the phyla of the bacteria. (E) “Small-network” identification based on a “smallworldness” index and the average shortest path length of the global bacterial community network in the air, marine, and soil environments. (F) Degree—the betweenness centrality plot of each node in the co-occurrence network. The nodes in red are viewed as keystone species. The size of the nodes shows the relative proportions of the OTUs in the total microbiome.
Study findings
There were 10,897 taxa detected from 370 individual air samples, and most bacterial sequences belonged to five phyla. Firmicutes, Alphaproteobacteria, Gammaproteobacteria, Actinobacteria, and Bacteroidetes constituted 24.8%, 19.7%, 18.4%, 18.1%, and 8.6% of these bacterial sequences, respectively. The abundance–occupancy relationship (AOR) between the samples a bacterial taxon occupied and its average mass in the global air showed a sigmoid curve, similar to the observed pattern for wild animals and plant distribution on Earth.
Air is a free-flowing, dynamic ecosystem enabling the long-range transport of bacterial communities it carries. However, its bacterial community appeared well connected to local environments, especially the source contributions and air quality conditions resulting from anthropogenic activities. Reduced environmental filtering effects and elevated human-related source contributions have led to fewer biomass loadings, higher pathogenic bacteria abundance, and more destabilized network structures.
Notably, compared to their counterparts in topsoil and marine environments, airborne bacteria were not closely interconnected, having an average intranode connection of 5.24. They had a random clustering approach, and the topology had low resistance to changes. The observed distant relationships and loose clusters of the network suggested that the airborne bacterial community was more liable to be perturbed as a function of environmental conditions that usually lead to drastic changes in bacterial composition. The functions of atmospheric bacterial taxa were inferred based on their genetic information in other habitats.
The team found potential associations between airborne bacterial communities and other surface microbial habitats. The estimated total abundance of global airborne bacteria (1.72 × 1024 cells) was comparable to those of the hydrosphere and one to three orders of magnitude lower than other habitats (e.g., soil).
Of the 23 major Earth habitats studied in the current study, terrestrial air exhibited more similarity to human- and animal environments, while offshore air showed a closer relationship to oceanic systems. Furthermore, evaluations based on Bayesian methods showed that the characteristics of the corresponding surface environment determined the dominant sources of airborne bacteria. Notably, human-related sources contributed more to the airborne bacteria in urban areas, especially on onshore sites, a finding that was majorly ignored in previous emission modeling studies.
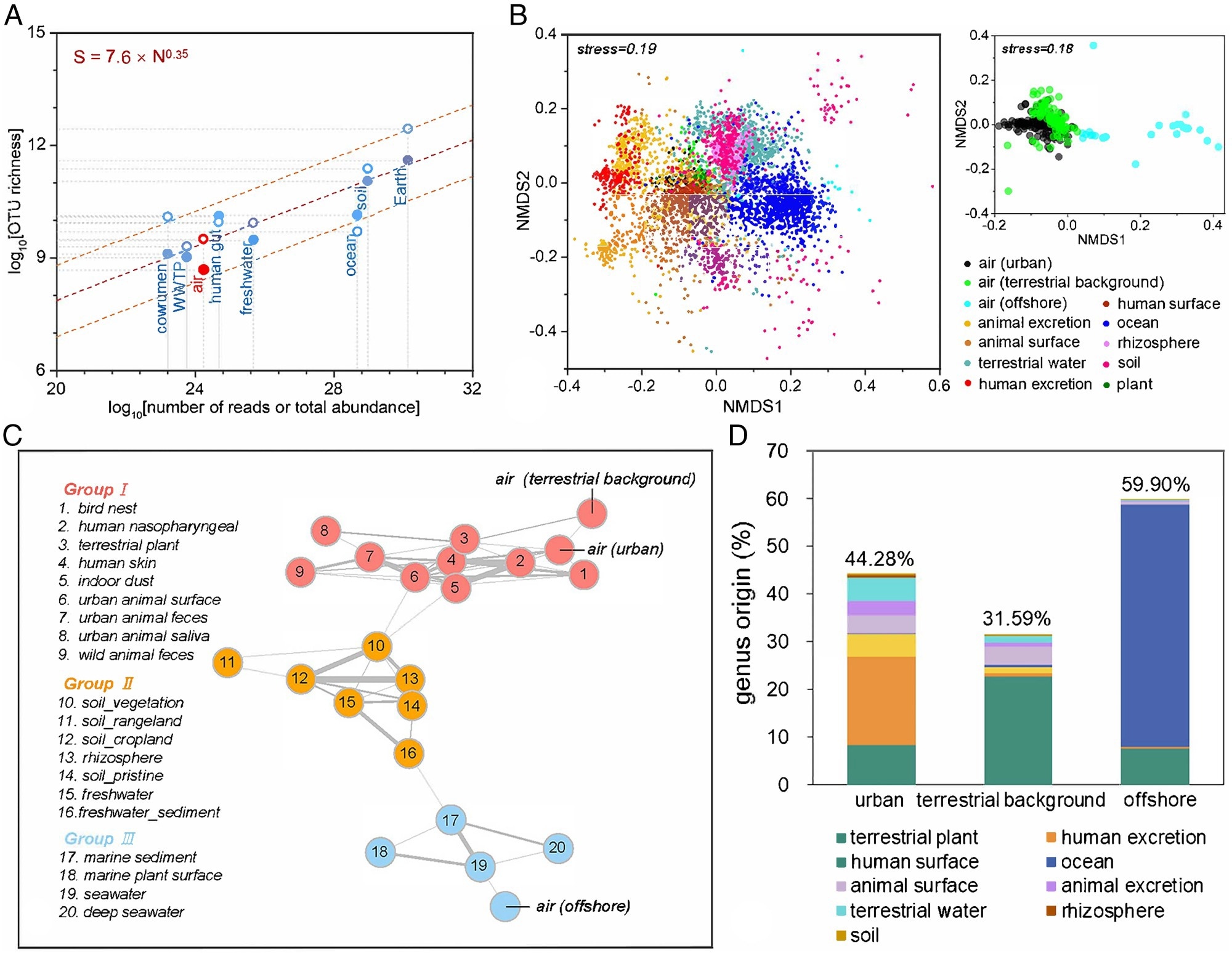 Role of airborne bacteria in the Earth’s microbial world. (A) Estimation of the global microbial abundance and richness in various habitats. The global richness (S) and the total abundance (N) in the corresponding habitats show a scaling relationship (the dashed orange line is the 95% prediction interval). Richness was predicted from the lognormal model using Nmax inferred from our sequencing data (filled circles) or Nmax predicted from the dominance-scaling law (open circles). The estimated S and N values for each habitat are, per see, a global sum. Some S and N were derived from previous studies. (B) A Bray–Curtis-based nonmetric multidimensional scaling (NMDS) plot shows that different microbial habitats harbor different bacterial communities on the Earth (n = 5,189). The Bray–Curtis distance was calculated to represent dissimilarities in the composition of bacterial communities. (C) Earth’s bacterial co-occurrence network shows the relationships of interconnection among 23 major microbial habitats. The connections (edges) stand for a strong (Spearman’s ρ > 0.7) and significant (p < 0.01) correlation. The thickness of lines represents the value of Spearman’s ρ. The environments were clustered into three groups with different colors by modularization. (D) Global airborne bacteria source analysis. Percentage of potential bacterial genera contributions from various environments to airborne bacterial communities in the urban, terrestrial background, and offshore areas, respectively, on a global scale.
Role of airborne bacteria in the Earth’s microbial world. (A) Estimation of the global microbial abundance and richness in various habitats. The global richness (S) and the total abundance (N) in the corresponding habitats show a scaling relationship (the dashed orange line is the 95% prediction interval). Richness was predicted from the lognormal model using Nmax inferred from our sequencing data (filled circles) or Nmax predicted from the dominance-scaling law (open circles). The estimated S and N values for each habitat are, per see, a global sum. Some S and N were derived from previous studies. (B) A Bray–Curtis-based nonmetric multidimensional scaling (NMDS) plot shows that different microbial habitats harbor different bacterial communities on the Earth (n = 5,189). The Bray–Curtis distance was calculated to represent dissimilarities in the composition of bacterial communities. (C) Earth’s bacterial co-occurrence network shows the relationships of interconnection among 23 major microbial habitats. The connections (edges) stand for a strong (Spearman’s ρ > 0.7) and significant (p < 0.01) correlation. The thickness of lines represents the value of Spearman’s ρ. The environments were clustered into three groups with different colors by modularization. (D) Global airborne bacteria source analysis. Percentage of potential bacterial genera contributions from various environments to airborne bacterial communities in the urban, terrestrial background, and offshore areas, respectively, on a global scale.
The authors noted no substantial disparities in the richness of airborne bacterial communities between urban and natural areas within the same latitude range. However, the geographic location did have a role to play. Thus, the evenness of the bacterial communities was much lower in urban air. For instance, the relative abundance of pathogenic species, Burkholderia and Pseudomonas, was higher in urban areas versus natural areas (5.56 and 2.50% vs. 1.44 and 1.11%). Furthermore, the bacteria contributed less to particulate matter (PM) mass in urban than in natural areas, indicating that urbanization increased the proportion of non-biological particulates in the air (e.g., dust).
The pathogens with the highest risk of mortality, Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (ESKAPE) were more abundant in urban air. The co-occurrence network of urban airborne bacterial communities indicated that anthropogenic impacts destabilized their network structure, which, in turn, also altered the bacterial taxonomic composition.
The authors noted that multiple factors impacted airborne bacterial communities—for instance, the geographic locations together with typical environmental factors. The biotic interactions among keystone and core bacterial communities, as well as bacterial richness, interacted significantly. Of all deterministic processes, environmental filtering was the primary determinant of the structure and distribution of airborne microbial communities.
Conclusions
To summarize, nearly 46.3% of the airborne bacteria originated from surrounding environments, and stochastic processes primarily shaped community assembly. Furthermore, the distinguishing feature of airborne bacteria in urban areas was its increasing proportion comprised of potential pathogens from human-related sources. Lastly, airborne bacterial source profiles affected a substantially higher percentage of the structural variations than that for air quality and local meteorological conditions (43.7% vs. 29.4% and 25.8%), as assessed via the variation partition analysis (VPA).
Journal reference:
- Global airborne bacterial community—interactions with Earth’s microbiomes and anthropogenic activities, Jue Zhao, Ling Jin, Dong Wu, Jia-wen Xie, Jun Li, Xue-wu Fu, Zhi-yuan Cong, Ping-qing Fu, Yang Zhang, Xiao-san Luo, Xin-bin Feng, Gan Zhang, James M. Tiedje, Xiang-dong Li, PNAS 2022, DOI: https://doi.org/10.1073/pnas.2204465119, https://www.pnas.org/doi/full/10.1073/pnas.2204465119
 New bacterial control strategy helps revive failing last-line antibiotics
New bacterial control strategy helps revive failing last-line antibiotics