In a recent study posted to the bioRxiv* preprint server, researchers in Italy performed a genome-based comparison of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) XBB recombinant with its parental lineages.
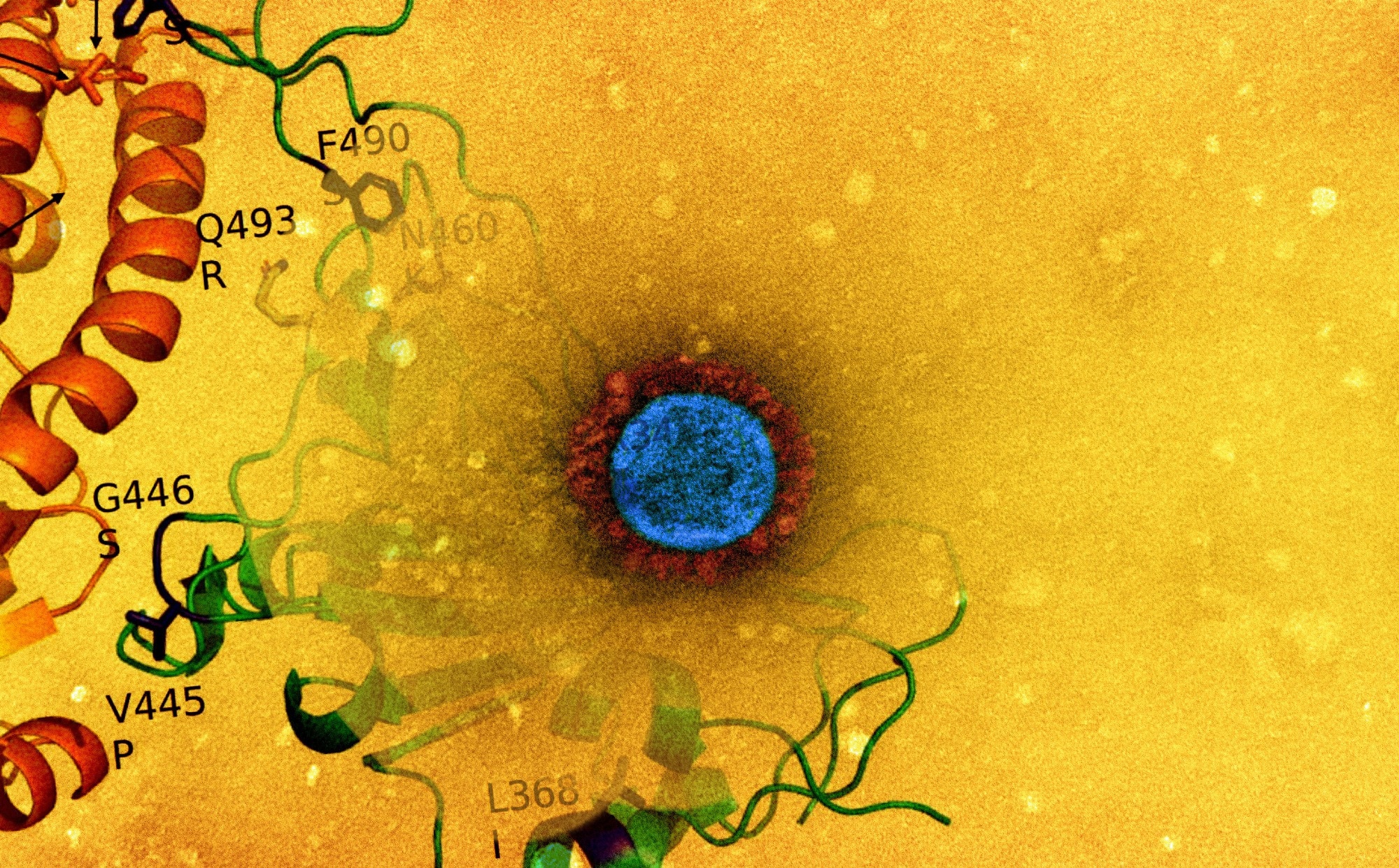 Study: Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. Image Credit: NIAID
Study: Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. Image Credit: NIAID
Background
The most recently detected SARS-CoV-2 recombinant is termed XBB lineage. The XBB lineage is a recombinant of the BA.2 lineage members BJ.1 and BM.1.1.1. According to the sequences reported to Global Initiative on Sharing All Influenza Data (GISAID) as of 2 December 2022, XBB and its descendants had a global sequence predominance of approximately 7%. The Technical Advisory Group on SARS-CoV-2 Virus Evolution (TAG-VE) highlighted the growing benefit of this sublineage, as well as some preliminary data on clinical severity and risk of reinfection in numerous countries.
Recent research suggests that XBB and XBB.1 can evade the antibody-mediated protection acquired by vaccination or prior infection. However, fully immunized individuals continue to be protected against hospitalization and mortality. XBB needs continual genome-based epidemiological surveillance as well as clinical management of illness features due to its highly immuno-evasive capabilities.
About the study
The present study conducted a genome-based survey to compare the novel SARS-CoV-2 recombinant XBB with its parental lineages.
To identify XXB and XXB.1 through an evolutionary standpoint, the genomic epidemiology related to SARS-CoV-2 Omicron variants was reconstructed with a global subsample collected over the last six months. In order to compare the genetic profile corresponding to XXB and XBB.1 and their ancestral lineages, the following subsets were created: BJ.1, BM.1.1.1, and XBB. Genetic analyses were conducted independently for the datasets. Genomes were arranged by employing the L-INS-I algorithm incorporated into Mafft 7.471. The evolutionary rate was determined using Bayesian Inference (BI) and the BEAST 1.10.4 program. Using the Bayes Factor test, the superior model was chosen to draw conclusions about the best representative output.
On a dataset sequence consisting of all studied genomes from the two parental lineages and the recombinant lineage, or BJ.1+BM.1.1.1+XBB, the breakpoint related to the recombination event was identified. The mutations defining the SARS-CoV-2 XBB and XBB.1 spike lineages were distinguished using consensus sequences which were obtained by applying a 75% threshold to all sequences available. After individuation, the mutations were confirmed using results from the GISAID web page titled "Lineage Comparison." Residue-residue interactions present on the interface were evaluated, and in-silico mutagenesis results were produced. Furthermore, the residues on the interface between SARS-CoV-2 receptor-binding domain (RBD) and angiotensin-converting enzyme-2 (ACE-2) were scanned for alanine.
Results
Reconstruction of the phylogenetic tree revealed that the XBB and XBB.1 genomes cluster inside the non-monophyletic GSAID Clade 21L. In particular, they are closely related to the BA.2 genomes, which constitute their ancestor. The Bayes Factor results corresponding to the BJ.1, BM.1.1.1, and XBB datasets demonstrated that the Bayesian Skyline Model concerning the relaxed clock model suited the data considerably better than the other clock and demographic models investigated for all datasets.
Bayesian Skyline Plot (BSP) associated with the recombinant XBB demonstrated that after the initial period of flattened genetic variability, there was an expansion in the size of the viral population beginning on 27 September 2022, reaching its peak almost on 6 October 2022. This was succeeded by a plateau phase that was observed up to a few days prior to 12 November 2022, when a decrease in viral population size was noted. After that, the number of lineages increased slowly until approximately 27 September 2022, after which the number of lineages ceased expanding.
BSP related to the sublineage XBB.1 exhibited a short phase of flattened genetic variability, followed by an increase in the viral population size, which began on 31 August 2022, attaining its peak on almost 10 September 2022. This phase was succeeded by a plateau that is currently ongoing and has exhibited no significant genetic variability or change in viral population size. Until around 6 September 2022, the number of lineages increased moderately slowly. Thereafter, the number of lineages ceased expanding.
Conclusion
The study findings showed that genome-based analysis of the SARS-CoV-2 XXB recombinant and its first sublineage XBB.1 suggested that, despite the number of spike mutations of interest possessed by the new variant, it currently lacks evidence of any specific virulence or high expansion capacity, despite its immuno-evasive properties. Nevertheless, its regional dominance has sparked anxiety, prompting the necessity for a thorough investigation. The data presented here indicate a period of rapid growth followed by a long phase of flattened genetic variability. This profile differs from an epidemiologically harmful lineage, as noted at the onset of the pandemic when the population size exhibited a steeply ascending curve.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference:
- Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages, Fabio Scarpa, Daria Sanna, Ilenia Azzena, Marco Casu, Piero Cossu, Pier Luigi Fiori, Domenico Benvenuto, Elena Imperia, Marta Giovanetti, Giancarlo Ceccarelli, Roberto Cauda, Antonio Cassone, Stefano Pascarella, Massimo Ciccozzi, bioRxiv 2022.12.20.521197, DOI: https://doi.org/10.1101/2022.12.20.521197, https://www.biorxiv.org/content/10.1101/2022.12.20.521197v1
 SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity
SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity