Over the past few centuries, the human population explosion and its associated need for livestock as food and labor to aid in the agricultural industry has resulted in the global upscaling of livestock rearing. The agricultural feedback loop of intensive farming systems enables larger livestock populations, which in turn demands increased crop production. This has resulted in livestock populations now exceeding the combined populations of humans and wild animals.
These practices, in combination with the long-distance transport of reared animals, have contributed to low-genetic diversity and high-density livestock. This presents an ideal recipe for outbreaks of pathogens capable of wiping out millions of livestock without the genetic capacity for resistance, which, when transported, can infect not only other livestock populations but also wild populations of the same or similar species.
Alarmingly, this cocktail of events is hypothesized to promote the emergence of novel zoonotic pathogens, arising from pathogens jumping to new hosts and mutations in previously benign microbiota previously associated with reared animals.
“This route to pathogen emergence may be particularly important in intensive farming systems, where large population size and high population density may select for traits associated with pathogenicity, while biosecurity reduces the risk of novel pathogens entering the population.”
Streptococcus suis is a ubiquitous microbiota component of the upper respiratory tract of pigs. Previously benign, intensive rearing of pigs in the 19th and 20th centuries, the microbe has been observed to adapt to a more pathogenic lifestyle. In 1954, the bacteria was implicated in widespread disease in pigs, and today presents one of the most common ailments in piglets. Alarmingly, accumulated mutations have allowed the bacteria to zoonotically spill over to humans, associated with meningitis, arthritis, endocarditis, and septicemia, with sudden death in both human and porcine hosts.
Following the first human S. suis-associated mortality in 1968, the bacteria has since led to large outbreaks in China and presents one of the leading causes of adult septicemia and meningitis across Southeast Asia.
“Difficulties in identifying the determinants of pathogenicity in S. suis have been attributed to its complex pathogenesis and high level of genetic diversity. Few studies have considered virulence factors in strains other than ST 1, which is responsible for most cases of S. suis disease in both pigs and humans worldwide.”
About the study
In the present study, researchers investigated the associations between intensive pig rearing, the emergence of novel S. suis lineages, and their potential for zoonotic spillover. They conducted a population genomic analysis of over 3,000 bacterial samples derived from tonsil and nasal swabs from pigs and wild boar. They additionally collected infected blood from humans and pigs suffering from S. suis disease across North America, Europe, Asia, and Australia. They aimed to elucidate the emergence, geographic spread, and degree of diversification of pathogenic lineages of the bacteria.
The study dataset comprised 3,070 genomic isolates of S. suis samples derived from previously existing published data and collected and sequenced as a part of this project. This included 29 published reference genomes and collection isolates from 15 countries spread across the five continents above. Sampling was conducted between 2014 and 2018. Isolates were processed through the Illunima whole genome HiSeq 25000 sequencing pipeline, which was then used to build a genomic library of isolates. Raw sequences were quality-checked, cleaned, and used to generate de novo assemblies for polymorphism evaluations.
The pipeline described by Athey et al. was used for serotyping and sequencing-typing analyses. Generated genomes were subsequently annotated to identify homologous genes and analyze pathogenicity-associated genomic islands. The PopPunk software was then used to identify divergent genomes and classify them into lineages. The six most common lineages thus identified were tested for temporal signals using a regression of root-to-tip distances against the year of isolate sampling. Finally, ancestral state reconstructions were used to infer the geographic spread of identified lineages.
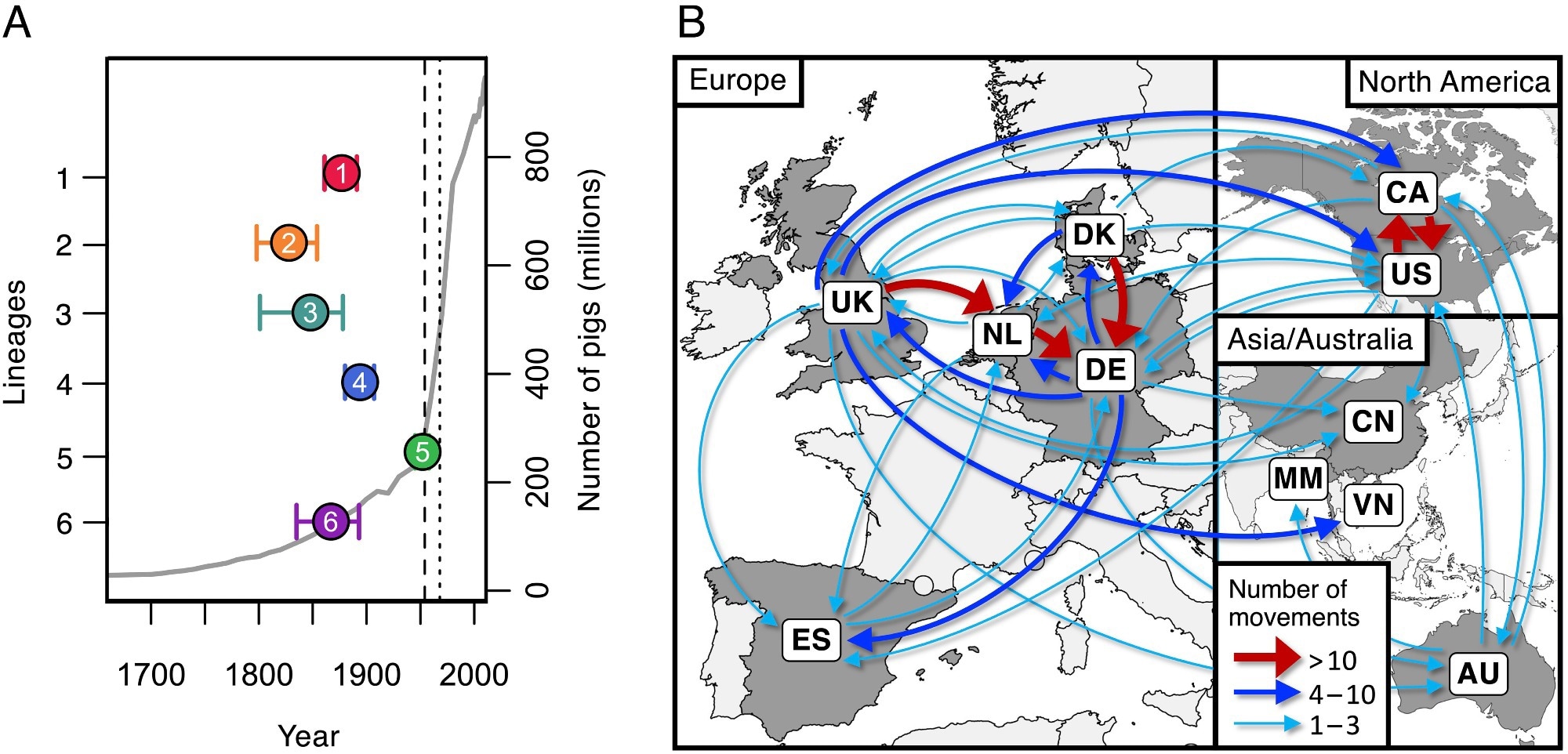 Dates of emergence and paths of between-country transmission for the six most common pathogenic lineages. (A) Estimates of the dates of the most recent common ancestors of the six most common pathogenic lineages (colored points) against an estimate of the global number of pigs (gray line). The vertical dashed line shows the date of the first reported case of S. suis disease in pigs (1954), and the dotted line shows the first reported human case (1968). (B) Map showing inferred routes of transmission of these six pathogenic lineages between the countries in our collection. Arrows represent routes with at least one inferred transmission event. Routes with more than ten inferred transmission events are shown in red, those with more than three in blue, and those with one to three in turquoise.
Dates of emergence and paths of between-country transmission for the six most common pathogenic lineages. (A) Estimates of the dates of the most recent common ancestors of the six most common pathogenic lineages (colored points) against an estimate of the global number of pigs (gray line). The vertical dashed line shows the date of the first reported case of S. suis disease in pigs (1954), and the dotted line shows the first reported human case (1968). (B) Map showing inferred routes of transmission of these six pathogenic lineages between the countries in our collection. Arrows represent routes with at least one inferred transmission event. Routes with more than ten inferred transmission events are shown in red, those with more than three in blue, and those with one to three in turquoise.
Study findings
Study findings revealed that over the past 200 years, rearing porcine populations have increased by over 200-fold, with the maximum increase during the latter half of the 20th century. These increases have resulted in the admittedly gradual yet concerning emergence of over 10 lineages with highly pathogenic life histories. The high density of reared pigs, usually antibiotic-treated, has led to diverse, antibiotic-resistance lineages of S. suis presenting significant control challenges.
Analyses of samples from Spain reveal that pigs, both reared and wild boar, are hosts to S. suis strains that are highly genetically diverse, suggesting that the association between the microbe and its host has been long-standing. Genetic dating analyses, however, revealed that all of the six most common pathogenic S. suis strains emerged during the 19th and 20th centuries, corresponding with the unprecedented increase in host rearing.
“The conclusion that these dates reflect an ecological shift toward pathogenicity in at least some of these lineages is supported by evidence that they coincided with the acquisition of a pathogenicity-associated genomic island (Island 3). It is further supported by patterns of genome reduction in each of the pathogenic lineages. In comparisons across bacterial species, it has been shown that bacterial pathogenicity is broadly associated with smaller genomes and fewer genes.”
Analyses of the metabolic capacities of pathogenic lineages revealed that at least two identified genomic islands had significantly upped their capabilities for within-host growth, with all six islands depicting increased metabolic activities over their more benign counterparts. In other bacteria studies, both within-host growth and metabolic rate have been linked to increased virulence, suggesting a trend in S. suis adapting to a more virulent life history with greater potential for zoonotic spillover.
“Pathogenic lineages may be better able to exploit particular regions of the tonsil than commensal lineages and vice versa, thereby reducing within-host competition. This could lead to segregation of these populations and reduced gene flow between them, which could in turn lead to the genome reduction in more pathogenic lineages due to fewer opportunities for gene acquisition from more diverse commensal lineages.”
Finally, analyses revealed that current S. suis strains depict a high rate of spread, capable of rapidly infecting an entire farm of pigs with the addition of one or a few infected individuals. The large-scale transport of livestock thereby presents an additional problem: previous endemic virulent strains being transmitted across nations or even continents, capable of infecting novel host populations with little to no innate resistance against them.
“Our results provide a framework for understanding the genomic diversity in S. suis and its association with pathogenicity. This is likely to be of widespread use in S. suis research and in informing strategies for controlling the burden of this disease on pig farming and human health. As our collection spans only a small proportion of the countries that farm pigs globally, further sampling from a broader range countries and more extensive sampling within countries, particularly those with large and growing pig populations, is needed to investigate the existence of additional pathogenic lineages that are geographically restricted or have recently emerged.”
 Pioneering gene therapy for rare immune disorder shows promise in early pre-clinical studies
Pioneering gene therapy for rare immune disorder shows promise in early pre-clinical studies