Researchers further used short tandem repeat sequencing at 50 known disease loci to understand human genomic diversity holistically and to pave the path for future studies in genomic medicine.
 Study: The landscape of genomic structural variation in Indigenous Australians. Image Credit: gopixa / Shutterstock
Study: The landscape of genomic structural variation in Indigenous Australians. Image Credit: gopixa / Shutterstock
The historic diversity of Australia
Australia is both a continent and an island, allowing it to form an isolated cradle for early man. Hundreds of Aboriginal clans and communities have been hitherto identified with potential genetic variations that allowed them to thrive in the continent's diverse environments over the last 50,000 years or more. Many of these peoples' cultural and linguistic history has been studied over the years, with over 250 languages described, 150 of which are still spoken today.
Unfortunately, despite intensive research on the cultural history of Indigenous Australians, their genomic diversity remains one of the most poorly understood of any community the world over. This knowledge gap is highlighted by the glaring lack of Indigenous Australian genomic data in the 1000 Genomes Project, the gnomAD reference database, or the Human Pangenome Reference draft genome. The databases are essential in current and future clinical interventions against genetic diseases. Therefore, the lack of Australian representation could significantly hamper genetic medicine efforts within the continent.
From the pure research perspective, Indigenous Australians represent almost a million humans or 1/8th of the global population. However, this proportion is both deceiving and an underrepresentation of the actual genetic diversity of these people, given their prolonged genetic seclusion from the rest of the world. Documenting the genomes of Indigenous Australians would allow for a better understanding of human genome evolution and the adaptations that allowed these remarkably resilient clans to persist in challenging environments, often without modern technological comforts upon which the rest of the world depends.
About the study
The present study forms a part of the National Centre for Indigenous Genomics (NCIG), a large-scale cohort aimed at revealing the genomics of Aboriginal Australians and Torres Strait Islander communities. The study cohort consisted of four NCIG-partnered Indigenous communities along with non-Indigenous Australians. The study methodology utilized long-read Oxford Nanopore Technologies (ONT) alongside the recently developed telomere-to-telomere human reference genome (T2T-chm13).
The four communities sampled included the Wurrumiyanga, Millikapiti, and Pirlangimpi tribes from the Tiwi Islands (NCIG-P1), the Galiwin'kus (NCIG-P2), the Titjikalas (NCIG-P3), and the Yarrabah (NCIG-P4). Sample cohorts were 9-41 in number (per community) for a total of 121 individuals. Additionally, 18 Australians of European descent were sequenced for comparison. Data collection was carried out between 2015 and 2019 and comprised the isolation of high molecular weight DNA from either blood or saliva samples. Between 10- and 30-fold DNA coverage was acquired from ONT sequencing of these samples.
Structural variants (SVs) obtained from this study were compared against the T2T-chm13 reference genome. Both long- and short-reads were compared, with the former presenting significantly higher mappability and coverage compared to the latter. Additionally, indels (insertion-deletion events) were compared against the reference genome, revealing 159,912 unique SVs and 136,979 indels. Finally, copy-number variants (CNV) were investigated against the reference genome, revealing 156 unique regions of variable CNVs across the 121-individual-strong cohort.
Study findings
This study surpassed the recently published ONT sequencing analysis of Icelanders as the largest discovery of novel SVs since the development of the Oxford Nanopore Technology. This is noteworthy given that the Icelander study included a 3,622-individual strong cohort, compared to only 121 individuals herein. This highlights the surprisingly high amount of genetic divergence of Indigenous Australians when compared to other historically isolated communities.
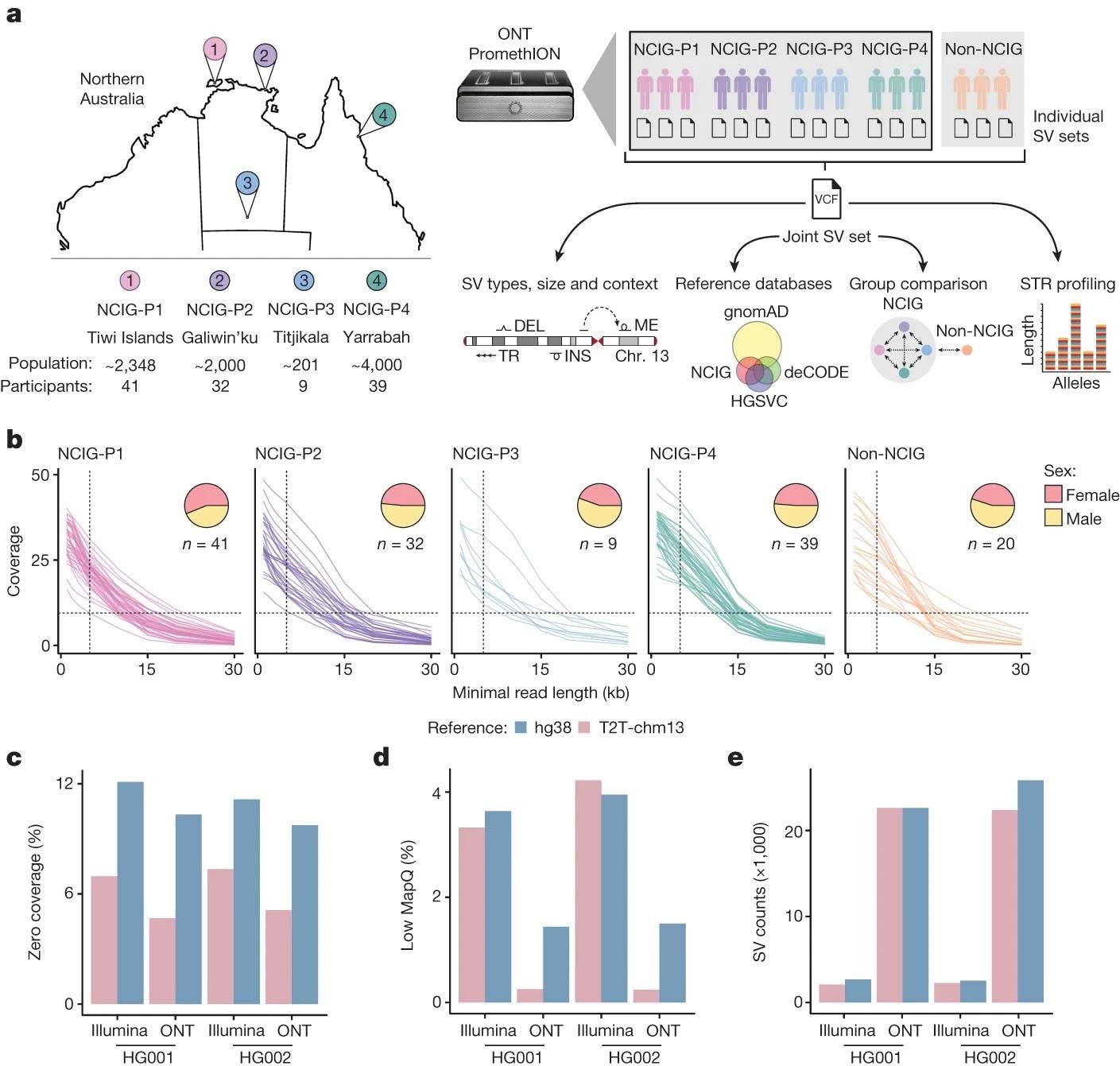
a, Study design and analysis workflow. DNA samples were collected from four Indigenous communities: Tiwi Islands (NCIG-P1), Galiwin’ku (P2), Titjikala (P3) and Yarrabah (P4), and from unrelated European individuals (non-NCIG). The map shows geographic locations, with population sizes and participant numbers underneath. ONT sequencing was performed and reads aligned to the T2T-chm13 genome. SVs were called for each individual, then joint calling was performed to generate a non-redundant set of SVs, genotyped for each individual. SVs were characterized by type, size and context and compared to existing SV datasets. SVs were compared between individuals and communities, with non-NCIG individuals as an outgroup. Short tandem repeat (STR) alleles were genotyped to assess variation. Chr, chromosome; DEL, deletions; INS, insertions; ME, mobile elements. b, Average genomic coverage as sequencing reads were filtered by a minimum read-length cut-off. Each line represents one individual. Pie charts show the proportion of male and female participants from each community. c, Percentage of genome with zero coverage for Illumina short-read and ONT long-read libraries from HG001 and HG002, aligned to either hg38 or T2T-chm13. d, Percentage of genome covered by alignments with low mapping quality (MAPQ < 5). e, Number of SVs detected.
Stratifications of SVs by size, type, and context revealed that 84.9% of all non-redundant variants were composed of repeats. Interestingly, despite being few in number, CNVs comprised >65 Mb of sequence data across the Australian cohort. Indel analyses revealed deletions of 13 Mb (average 243 kb) and gene duplications of 1.8 Mb (average 303 Kb). Most of the observed variation was concentrated around the telomeric ends of the Australians' chromosomes. Comparisons with the T2T-chm13 reference (European origin) suggest that most of the SVs in Indigenous Australians arose due to transposition events (jumping genes following gene duplication).
"Given the inclusion of unique, under-represented Australian communities and the use of long-read sequencing, our catalog contained a high proportion of SVs that have not been previously annotated"
Analyses of Indigenous genomes versus the reference suggest that between 19-62% of the Australian genome (specifically, regions of interest called in this study – SVs) is unique and novel to science. Distribution and diversity analysis conducted herein highlights the importance of large-scale sampling – 26.3% of unique SVs were found in a single individual, 65.6% were found in less than 50% of the population, and only 0.2% were observed across the entire sampled cohort.
"The clear genetic distinctions between Indigenous Australian and non-Indigenous Australian individuals was further reiterated by principal coordinate analysis (PCOA) and fixation index (FST) analysis of structural variation."
The generation of discovery curves for novel SVs revealed that many SVs could potentially exist in the population but were not included in the cohort due to the relatively small size (121 individuals sampled out of nearly 1 million). Highlighting these findings, the Yarrabah (NCIG-P4) community was found to have higher genomic diversity than the other three communities combined and by far the highest proportion of unique SVs.
The functional genetic disease context analysis identified an SV in the Galiwin'ku community (NCIG-P2) directly associated with the Machado–Joseph Disease (MJD), a late-onset movement disorder estimated to affect 5 out of every 100,000 people. Anecdotal evidence suggests that prevalence among Northern Territory Indigenous Australians is closer to 5 out of every 1,000 people, but hitherto, the reason for this 100-fold higher prevalence remained unknown.
These findings "prompted an ongoing dialogue between NCIG, Galiwin'ku representatives, local genetic counsellors and the MJD foundation (https://mjd.org.au/), who work with remote Northern Territory Aboriginal communities to develop unique clinical genetics service models tailored for their needs"
Conclusion
The current study marks the first and most comprehensive genomic study of the genetic diversity present within Aboriginal Australian communities. Analyses of high-resolution DNA from 121 individuals spread across four communities revealed almost 160,000 unique SVs and 137,000 indels, the highest in any genomic study of isolated communities to date. These findings highlight the substantial genomic diversity of this group, most of which was potentially missed in the 121-strong sample cohort.
The study helped explain the genetic underpinning governing the 100-fold higher prevalence of MJD in Northern Australia compared to the rest of the world, thereby highlighting the importance of including genomic data from isolated communities in reference genome databases, especially in the primary purpose of those databases – clinical interventions against genetic diseases – is to be fulfilled.
 Pork supply chains may be shaping the global spread of drug-resistant Salmonella
Pork supply chains may be shaping the global spread of drug-resistant Salmonella