An international collaboration of scientists from the CEA, CNRS, University Joseph Fourier, the EMBL and the ILL has revealed the molecular function of a protein essential for replication of H5N1 influenza virus. A sub-domain of the viral polymerase undergoes large-scale structural reorganization to enable an essential part of the polymerase to enter the nucleus of the infected cell, where the viral genome is replicated. This study, published in the Journal of the American Chemical Society, illustrates how the flexibility of a protein allows it to adapt its function, facilitating infection of the host.
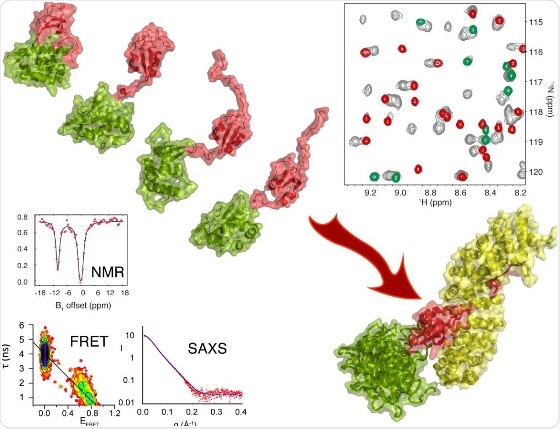
The structure of the 627-NLS domain (green and red) of the PB2 domain of H5N1 viral polymerase opens and closes at a rate of around 50s-1 at room temperature. The open form interacts with Importin α (yellow) allowing transport of this part of the polymerase into the nucleus of the cell. These results were derived from a combination of NMR (top right and left), single molecule FRET (bottom left); and small angle X-ray and neutron scattering (bottom). © Martin Blackledge
The polymerase allows the virus to reproduce copies of its genomic material in the infected cell, and thereby produce new viruses. It is known that adaption of the influenza virus occurs through mutations in the viral polymerase, in particular in the C-terminal domain 627-NLS of the PB2 polymerase protein. This two-domain protein is required for import of the viral polymerase into the nucleus, by binding to importin α.
The protein has been crystallized, in isolation and in the context of the entire polymerase, but the crystallized conformation appeared incapable of binding to importin α, due to a strong steric clash between importin α and 627-NLS. The molecular basis of this essential interaction therefore remained mysterious.
Researchers at the IBS, and their collaborators were able to show for the first time, using nuclear magnetic resonance spectroscopy (NMR), small angle scattering (SAS) and single molecule Förster resonance energy transfer (FRET), that in solution the protein exhibits a far more complex behaviour. In fact the conformation described by crystal structure indeed exists in solution, but this conformation exchanges, around 50 times per second, with another form of the protein, in which the two domains, attached by a flexible linker, dislocate and can move quite freely relative to each other.
Crucially, they were able to show that this 'open' form of the protein indeed interacts with importin α and it is this conformational equilibrium that allows for PB2 to enter the nucleus. The exchange between 'open' and 'closed' forms of the protein was also shown to be highly temperature dependent, and it seems possible that the equilibrium between closed and open conformations plays a role as a molecular thermostat, controlling the efficiency of viral replication in the different species where the virus needs to evolve as a function of the temperature of the host environment.
This study again highlights the remarkable efficiency of viruses to exploit conformational flexibility to extend their functional diversity with limited genetic material. The same two-domain protein has two distinct functions: The closed form of the protein is necessary for viral replication within the polymerase once the protein enters the nucleus, but the 'open' form is necessary for import into the nucleus. More generally, inter-domain dynamics play crucial roles in a multitude of molecular recognition, transport and signaling processes. These complex dynamic modes cannot be understood from static structures of either the entire protein or individual domains. The study demonstrates the importance of solution-state structural biology to accurately describe the relationship between structure, dynamics, thermodynamics and biological function.
 Excess tubulin disrupts tissue organization and cell survival
Excess tubulin disrupts tissue organization and cell survival