 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Background
The ongoing pandemic caused by SARS-CoV-2 has infected more than 311 million people worldwide and caused over 5.5 million deaths so far. Although the rapid development and distribution of coronavirus disease 2019 (COVID-19) vaccines have effectively reduced severe SARS-CoV-2 outcomes, large numbers of people have yet to be vaccinated due to inequities in vaccine access in many parts of the world.
Therefore, new research on genomics, molecular structure and dynamics, mechanisms of binding, and the life cycle of the SARS-CoV-2 infection is necessary to find effective COVID-19 treatment strategies.
About the study
In the present study, the researchers evaluated different antigen-antibody (Ag-Ab) complex interactions using antibody design software followed by constant-pH Monte Carlo (MC) calculations, constant-charge coarse-grain (CG) molecular dynamics (MD) calculations, and constant-charge atomistic MD simulations. These studies were conducted in an effort to identify the Ab candidate with comparatively higher affinity in all four types of evaluation using an existing experimental RBD-CR3022 complex as a template.
The initial screening was performed using ROSETTA-designed antibody structure software. According to its scoring function, the researchers ranked 235,000 candidates, of which only 383 candidates showed improved affinities in comparison to the CR3022-RBD complex.
From the 383 candidates, the researchers selected ten Ag-Ab complexes that showed the best affinity in all theoretical calculations. The ten selected candidates were further evaluated by free energy (ΔG) calculations using umbrella sampling (US) and FORTE.
Study findings
The free energy calculations of the RBD-Ab complex of the ten selected candidates using the umbrella sampling method showed that P01, P05, P06, P09, and P10 had significantly improved binding affinity as compared to CR3022. The free energy value was -18.3 kcal/mol for P01 and -7.1 kcal/mol to -10.8 kcal/mol for the P01-P05-P06-P09-P10 group relative to the wild-type CR3022 antibody. P01 displayed the best improved binding affinity in comparison to the native CR3022 and the P05, P06, P09, and P10 set.
The RBD-Ab binding free energy calculations through FORTE for the selected ten candidates were observed to be in the order of P01/P06 > P02/P05/P08/P09/P10 > P04 > P03 > P05. This indicated P01 and P06 as the two best candidates with free energy values of -0.788 KBT and -0.790 KBT, respectively.
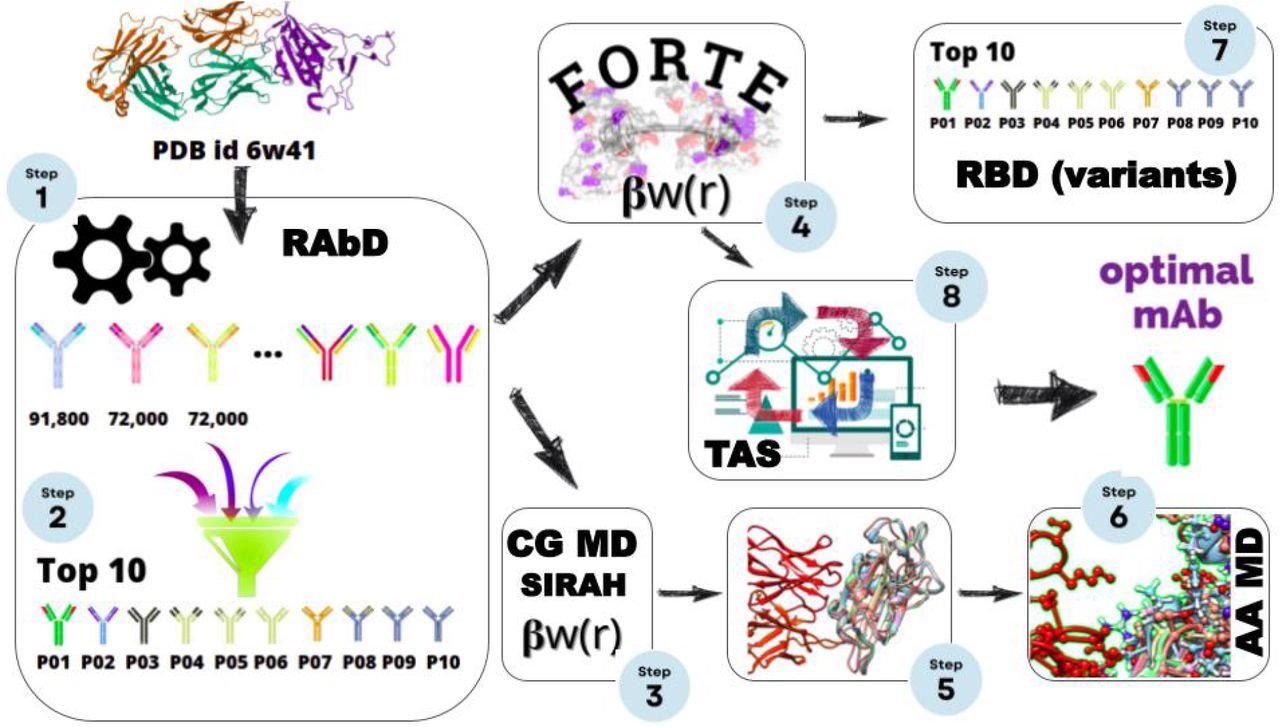
Scheme for the multiple scales in silico protocol, consisting of an initial structural-bioinformatics-based methodology to explore macromolecules as potential candidates (steps 1 and 2), constant-charge CG MD (steps 3, and 5), a constant-pH CG MC simulations (steps 4, 7, and 8), and an atomistic constant charge MD simulation. At the end of this cycle, an optimized mAbs with a higher binding affinity is obtained.
The researchers expanded the analysis and tested the binding affinities of these candidates with ROSETTA-designed fragments of mAbs with different SARS-CoV-2 variants. The results revealed an improved binding affinity for all ten candidates when compared with native CR3022. This indicates that all molecules from P01 to P10 can potentially block the interaction between SARS-CoV-2 and the host cell by preventing the RBD from being available for angiotensin converter enzyme 2 (ACE2) binding.
Interestingly, both the native CR3022 and all ROSETTA-designed binders displayed a higher affinity for the RBD of the SARS-CoV-2 Omicron variant, thus suggesting that CR3022 and mAbs derived from it may potentially neutralize the Omicron variant.
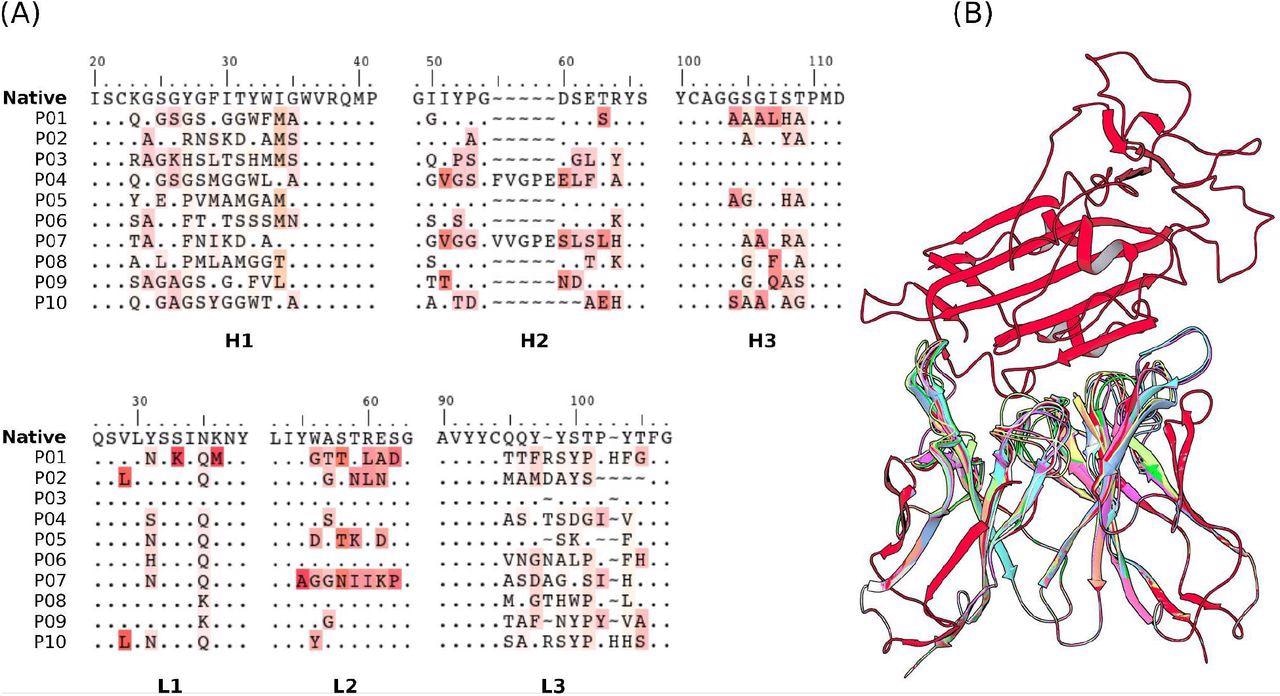
Sequences of the CDRs (A) and the structures (B) of the 10 selected mAbs for evaluations using US/SIRAH and FORTE methods. Dots represent identities. Residue letters in (A) were colored according to similarity with their counterparts in the reference native CR3022 sequence. In (B) RBD (up) was colored in red and each candidate antibody (down) in a different color.
The researchers further evaluated the contribution of a specific amino acid to the RBD binding of the two best binders, P01 and P06. This was achieved through the use of theoretical alanine scanning (TAS), in which an amino acid from the potential candidate was replaced by alanine and then repeated the complex simulation for this new possible binder.
The results of this experiment showed that amino acids located at the Ag-Ab interface and deep inside the protein structure are critical for the complexation process. This is because of the long-range nature of the electrostatic interactions and the electrostatic coupling between titratable groups, which are characterized as “electrostatic epitopes.”
The researchers also observed that the total net charge of the mAbs can impact binding affinity. To this end, they found a clear linear tendency for mAbs with low charges to exhibit higher binding affinity to RBD. The association of SARS-CoV-2 antigens with the studied mAbs was derived by Coulombic force, which was also observed in previous studies.
Conclusions
Overall, the multi-scale approach used in the present study is a fast, robust, and reliable tool to design better macromolecular ligands to identify the best mAb candidate for SARS-CoV-2 variants, including the Omicron variant. This multi-scale in silico approach is, therefore, a sensible and realistic tool for SARS-CoV-2 diagnosis, treatment, and prevention.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Neamtu, A., Mocci, F., Laaksonen, A., & Barroso da Silva, F. (2022). Towards an optimal monoclonal antibody with higher binding affinity to the receptor-binding domain of SARS-CoV-2 spike proteins from different variants. bioRxiv. doi:10.1101/2022.01.04.474958. https://www.biorxiv.org/content/10.1101/2022.01.04.474958v1.full.
- Peer reviewed and published scientific report.
Neamtu, Andrei, Francesca Mocci, Aatto Laaksonen, and Fernando L. Barroso da Silva. 2023. “Towards an Optimal Monoclonal Antibody with Higher Binding Affinity to the Receptor-Binding Domain of SARS-CoV-2 Spike Proteins from Different Variants.” Colloids and Surfaces B: Biointerfaces 221 (January): 112986. https://doi.org/10.1016/j.colsurfb.2022.112986. https://www.sciencedirect.com/science/article/pii/S0927776522006701.
 Scientists discover protective antibodies with potential to treat West Nile virus
Scientists discover protective antibodies with potential to treat West Nile virus