Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causal agent of the coronavirus disease 2019 (COVID-19) pandemic, is a highly pathogenic coronavirus belonging to the betacoronavirus genus.
The genome of SARS-CoV-2 consists of a single-stranded RNA of 29,903 nucleotides. SARS-CoV-2 is associated with a very high mutation rate, and, recently, machine learning has proved to be a valuable method to identify the distinctive genomic signatures among viral sequences. This could be helpful in taxonomic and phylogenetic studies and also help in detecting emerging variants of concern. In a new study posted to the bioRxiv* preprint server, researchers studied KEVOLVE, an approach based on a genetic algorithm with a machine learning kernel, to identify several genomic signatures.
 Study: Machine learning-based approach KEVOLVE efficiently identifies SARS-CoV-2 variant-specific genomic signatures. Image Credit: Metamorworks / Shutterstock
Study: Machine learning-based approach KEVOLVE efficiently identifies SARS-CoV-2 variant-specific genomic signatures. Image Credit: Metamorworks / Shutterstock
 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Machine Learning Method: KEVOLVE
KEVOLVE includes a machine learning kernel and is based on a genetic algorithm. It identifies minimal subsets of discriminative motifs. In the context of HIV, KEVOLVE-identified motifs facilitated the construction of models that out-performed specialized HIV prediction tools, thereby demonstrating the potential of this approach.
In the current study, researchers evaluated the KEVOLVE, whose search function was upgraded to identify smaller sets of motifs. It was important to maintain the same discriminative performance criteria. Scientists compared several reference tools to identify discriminating motifs among SARS-CoV-2 genome sequences. Four main steps were followed: (i) identification of motifs in a restricted set of nucleotide sequences, (ii) using the motifs to build prediction models and assessing them using a large set of SARS-CoV-2 sequences, (iii) analyzing the KEVOLVE-identified motifs to highlight their potential biological functions, and (iv) dedication of a specific analysis to the new Omicron variant.
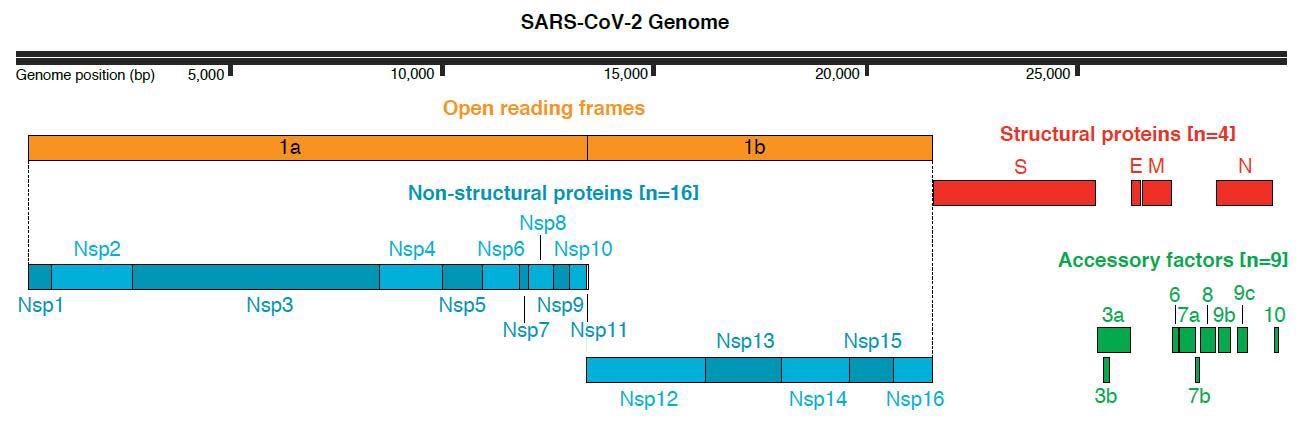
SARS-CoV-2 genome organization
Key Findings
In a comparative study in which scientists analyzed a large SARS-CoV-2 genome dataset, it was observed that KEVOLVE performed better in identifying variant-discriminative signatures when compared to several gold-standard reference statistical tools.
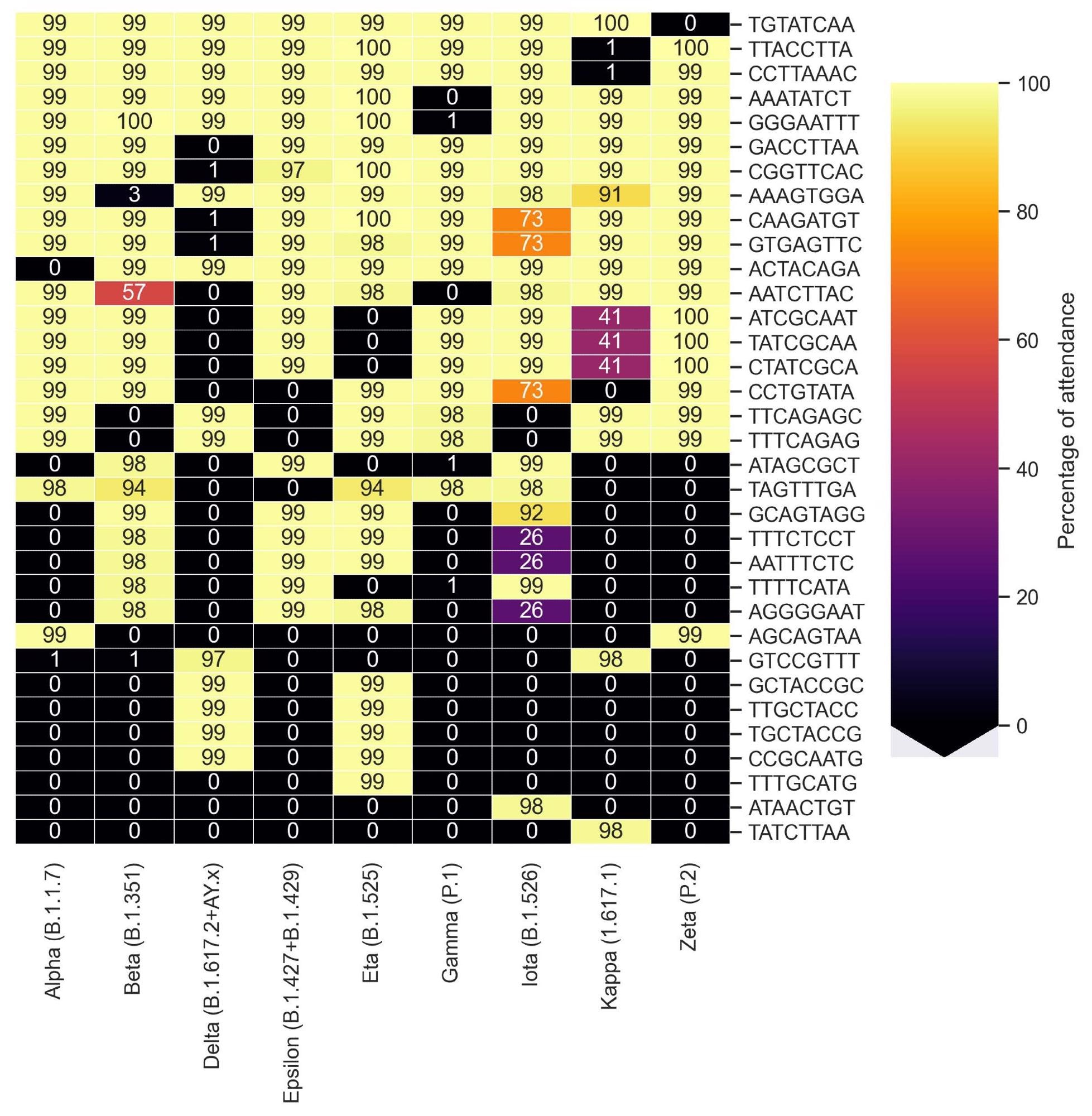 Cluster map representing the percentage of presence of motifs identified by KEVOLVE according to the groups of variants of SARS-CoV-2.
Cluster map representing the percentage of presence of motifs identified by KEVOLVE according to the groups of variants of SARS-CoV-2.
Next, the variant-discrimination motifs identified by KEVOLVE were analyzed to assess the potential functional impact of these mutations. The divergence between the genomes of SARS-CoV-2 variants was observed to be less than 1%, and the mean divergence between all the sequences was 0.29%. Omicron was observed to be the most divergent (0.44%), compared to other variants, such as Alpha, Zeta, and Iota. Overall, the variant-discriminative signatures were associated with known mutations among the different variants regarding the functional and pathological impacts based on the existing literature.
Using KEVOLVE, researchers were able to highlight three substitutions constituting unique features of Omicron: I3758V in ORF1ab (NSP6) and N679K and D796Y in ORF2. They stated that the functional implications of these mutations are unknown. Future research could investigate how these mutations could influence viral fitness and susceptibility to natural and vaccine-mediated immunity. It must be noted that the combination of N679K with H655Y and P681H could increase the cleavage of spike and, thereby, enhance fusion and viral transmission.
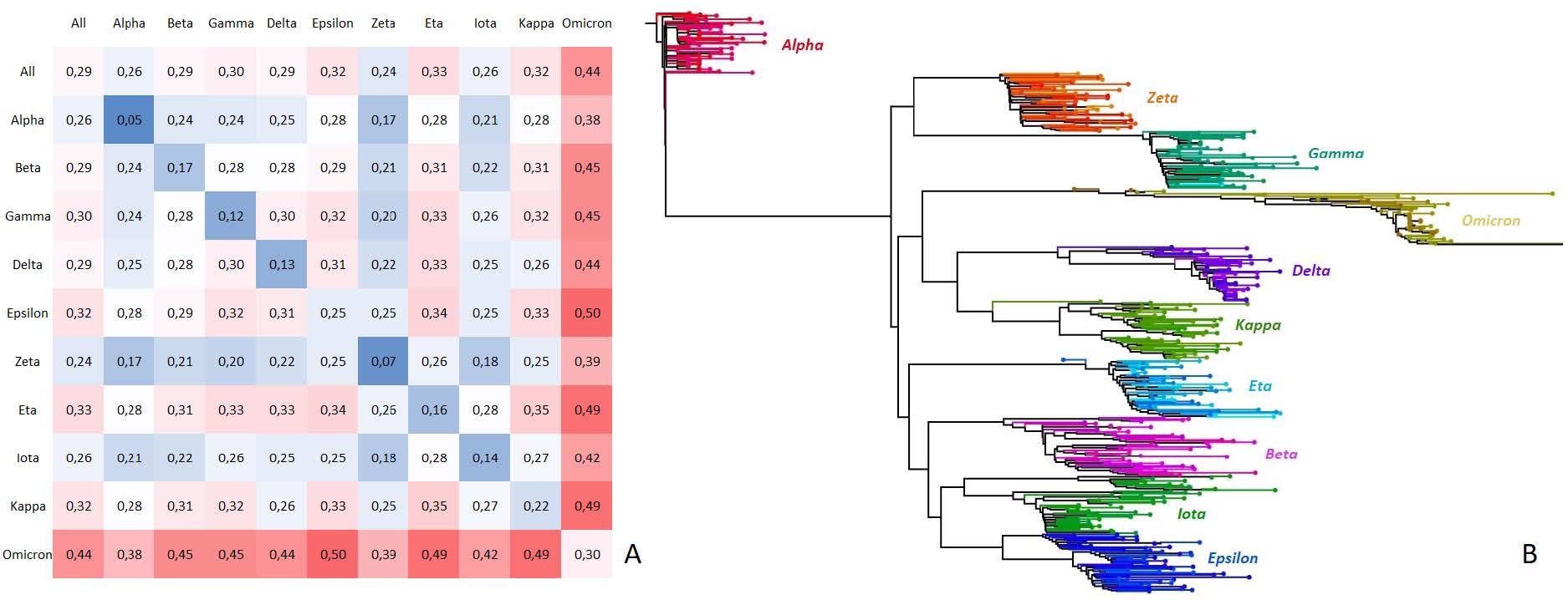
Nucleotide rate dissimilarity matrix and phylogenetic tree of SARS-CoV-2 variant families.
Implication of Results
The findings documented in the study suggest that KEVOLVE is a robust tool for the rapid and precise determination of SARS-CoV-2 variants. The genomic signatures could be used to build peptide or oligonucleotide libraries for quick pathogen detection using existing tools. Contrary to traditional methods, KEVOLVE is automatic and independent of multiple sequence alignments, a huge advantage. KEVOLVE also possesses the ability to be adapted to allow the automated analysis of previously-identified motifs, thereby increasing its efficiency even further.
Concluding Remarks
In the current study, researchers demonstrated the numerous advantages of machine learning-based tools over conventional methods to efficiently discriminate between SARS-CoV-2 variants. The new approach does not depend on multiple sequence alignment and enables users to capture mutations associated with motifs of interest. Furthermore, this detection could be done in different groups of viral pathogens.
Such methods could well be fundamental in the future to identify novel motifs pointing toward unrecognized mutations, of functional importance, in novel emerging variants. Scientists stressed that KEVOLVE could be a valuable complement to conventional genomic analyses to classify and understand viral variants.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 New AI tools enhance diagnosis and management of kidney disease
New AI tools enhance diagnosis and management of kidney disease