The ongoing COVID-19 pandemic is a leading cause of hospital admission and mortality worldwide despite the availability of many severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccines. Before December 2021, no therapies were available to treat SARS-CoV-2 in community settings. Hence, there was an urgent need for potent and safe oral antivirals for SARS-CoV-2 treatment, especially for those with a high risk of severe disease.
Nirmatrelvir (PF-07321332) is the first orally bioavailable antiviral agent which can inhibit the SARS-CoV-2 main protease (Mpro) enzyme activity and antiviral activities across a large spectrum of CoVs. Therefore, on 22 December 2021, the United States (US) Food and Drug Administration (FDA) issued emergency use authorization (EUA) for nirmatrelvir/ritonavir in SARS-CoV-2 treatment.
In preclinical studies, nirmatrelvir has demonstrated a benign safety profile and potent antiviral activity. However, it is unknown if nirmatrelvir maintains concentrations above the in vitro 90% effective concentration (EC90) in humans.
About the study
In the present accelerated randomized, placebo-controlled, and double-blind phase one study, the team of researchers evaluated the tolerability, safety, and pharmacokinetic profile of the oral antiviral agent, nirmatrelvir against SARS-CoV-2 with and without a pharmacokinetic enhancer drug named ritonavir in healthy adults.
The team utilized an innovative and seamless functional strategy integrating a flexibly written protocol that permitted the study to adapt to emerging information within the study without requiring a protocol revision.
Two interlacing single-ascending dose (SAD) study groups were assessed in a three-period crossover. Multiple-ascending dose (MAD) with nirmatrelvir/ritonavir two times daily (BID) dosing was evaluated for 10 days in five similar cohorts. The safety of the nirmatrelvir/ritonavir regimen was evaluated in SAD, MAD, and a supratherapeutic exposure cohort.
The integration of simulations and modeling of SAD and MAD data with non-clinical information and a quantitative systems pharmacology model (QSP) supported determining the dose and dosing regimen of nirmatrelvir/ritonavir for clinical efficacy evaluation in phase two and three clinical trials.
Study findings
The results indicate that the nirmatrelvir and ritonavir combination was safe and well-tolerated in supratherapeutic exposure, MAD, and SAD cohorts. All treatment-related adverse events (ADEs) were mild in severity, such as dysgeusia, mild elevation of thyroid-stimulating hormone (TSH), and nausea, and no severe ADEs were documented.
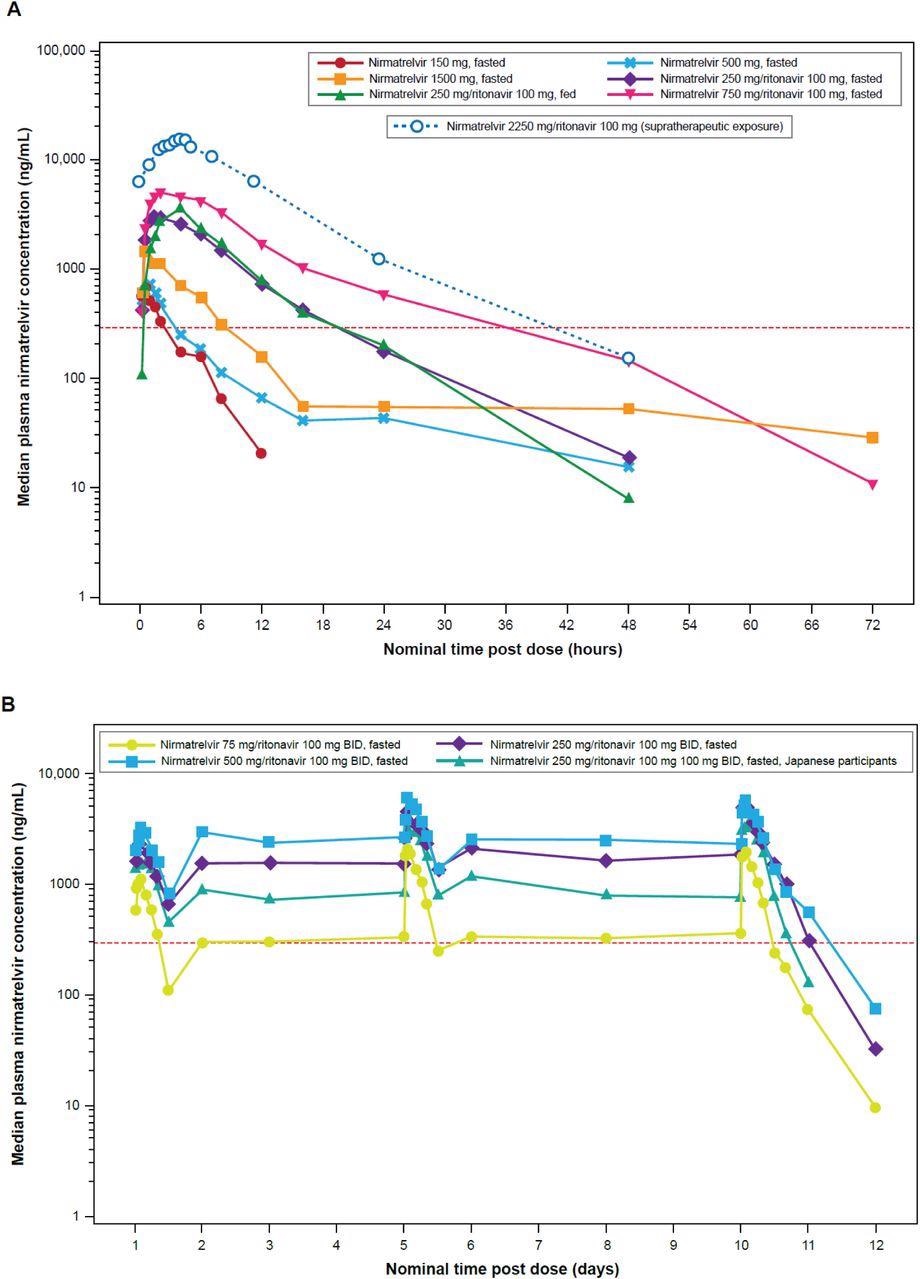
Median plasma nirmatrelvir concentration-time profiles (semi-log scales) for single-ascending dose and supratherapeutic exposure cohorts (A) and multiple-ascending dose cohort (B). For summary statistics, values below the lower limit of quantification (10 ng/mL) were set to zero. In the supratherapeutic exposure assessment, nirmatrelvir was administered as 3×750 mg doses at 0, 2, and 4 hours. In the single-ascending dose assessments where applicable and supratherapeutic exposure assessments, ritonavir was dosed at 100 mg at –12 hours, 0 hours, and 12 hours after dosing. In the multiple-ascending dose assessment, ritonavir was dosed at 100 mg twice daily. The red dotted line is EC90 of 292 ng/mL. BID=twice daily; EC90=concentration at which 90% inhibition of viral replication is observed.
The coadministration of ritonavir with nirmatrelvir increased the nirmatrelvir concentration by approximately eight times. The half-life and exposure of nirmatrelvir increased considerably by ritonavir, facilitating the selection of 300/100 mg BID dosage regimen of nirmatrelvir/ritonavir combination for phase two and three trials to attain free concentrations of nirmatrelvir above EC90 that are required for 90% inhibition of SARS-CoV-2 replication in vitro.
Since the effect of food on nirmatrelvir/ritonavir was minimal, the combination can be administered with or without food. Further, the QSP model indicated that a five-day nirmatrelvir/ritonavir 300/100 mg BID dosage regimen would substantially lower the SARS-CoV-2 load in COVID-19 patients with symptomatic disease and avert the development of the severe form of the disease, requirement of hospital admission, and mortality. Hence, supporting this dosing duration in phase two and three clinical trials. Nirmatrelvir and ritonavir 300/100 mg BID dosage regimens beyond five days did not demonstrate any therapeutic benefits in those with symptomatic COVID-19.
Conclusions
The study findings show that the nirmatrelvir and ritonavir combination was well-tolerated and safe in healthy subjects. Ritonavir stimulated the pharmacokinetics of nirmatrelvir and thus anticipated to attain plasma concentrations and maintain minimum plasma concentration (Cmin) values several times greater than the in vitro EC90 in more than 90% of the future subjects when dosed twice daily at a steady state.
This first-in-human study utilizing a novel, seamless, and efficient approach conducted in response to the urgent requirement of a COVID-19 treatment option enables the selection of dose and duration of nirmatrelvir/ritonavir for use in the second and third phases of clinical trials within six weeks of the first dose in humans along with crucial pharmacokinetic and safety assessment data to initiate pivotal efficacy trials. The present findings significantly lowered the overall development and decision timelines associated with the use of nirmatrelvir/ritonavir combinations in SARS-CoV-2 therapy.
One limitation of the study was the small size and limited ethnic diversity of the study population. Extensive studies with diverse patient populations evaluating the safety and efficacy of nirmatrelvir/ritonavir are either ongoing or completed.
Overall, the study reported pharmacokinetics and safety data to support using nirmatrelvir 300 mg combined with ritonavir 100 mg regimen administered two times daily for five days in the late phases of clinical trials COVID-19 patients.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Innovative Randomized Phase 1 Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir, Ravi Shankar P. Singh, Sima S. Toussi, Frances Hackman, Phylinda L. Chan, Rohit Rao, Richard Allen, Lien Van Eyck, Sylvester Pawlak, Eugene P. Kadar, Frances Clark, Haihong Shi, Annaliesa S. Anderson, Michael Binks, Sandeep Menon, Gianluca Nucci, Arthur Bergman, medRxiv 2022.02.08.22270649; doi: https://doi.org/10.1101/2022.02.08.22270649, https://www.medrxiv.org/content/10.1101/2022.02.08.22270649v1
- Peer reviewed and published scientific report.
Singh, Ravi Shankar P., Sima S. Toussi, Frances Hackman, Phylinda L. Chan, Rohit Rao, Richard Allen, Lien Van Eyck, et al. 2022. “Innovative Randomized Phase 1 Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID‐19 Trial of Nirmatrelvir.” Clinical Pharmacology & Therapeutics, April. https://doi.org/10.1002/cpt.2603. https://ascpt.onlinelibrary.wiley.com/doi/10.1002/cpt.2603.
 Fluoride myths and DIY dentistry dominate oral health misinformation
Fluoride myths and DIY dentistry dominate oral health misinformation