Neurons are the anatomical and functional unit of the nervous system, regulating both vital functions and higher functions. Among the very few cells in the body that do not regenerate, these cells must remain healthy despite numerous threats to their integrity, while still functioning normally. A new research paper in the journal EMBO Reports describes how DNA damage can affect neural health and function.
Neurons have highly efficient mechanisms to resist changes to the basic genetic code, called the DNA damage response (DDR) pathways. They suffer the most if loss-of-function mutations occur in these pathways, because of the many tasks they do that consume high amounts of energy besides requiring transcriptional accuracy.
If these pathways fail to kick in when required, deleterious genomic rearrangements, dysregulation of transcriptional pathways and the build-up of damaged genomic loci lead to cumulative toxicity to the neurons. Eventually, these result in cellular apoptosis, senescence, or unregulated cell division, marks of aging, and deterioration.
Neurodegenerative disease is the result of non-viable neurons, which in turn cause memory loss, motor impairment, and the loss of independence over the long run. Conditions associated with neurodegeneration are No. 3 on the causes of death in the USA, and No. 5 worldwide.
Recent research suggests that the vast array of DDR pathways play a significant role in preventing genomic DNA damage from both external injury and toxic processes within the body itself. Each pathway responds to a specific type of injury, using its own cascade of enzymes and repair proteins to first detect the lesion, and then either repair or excise the injured locus via nuclease activity before finally building in new DNA to fill in the gap with the help of a polymerase enzyme. The cut ends of the DNA strand are then sealed together using a ligase enzyme.
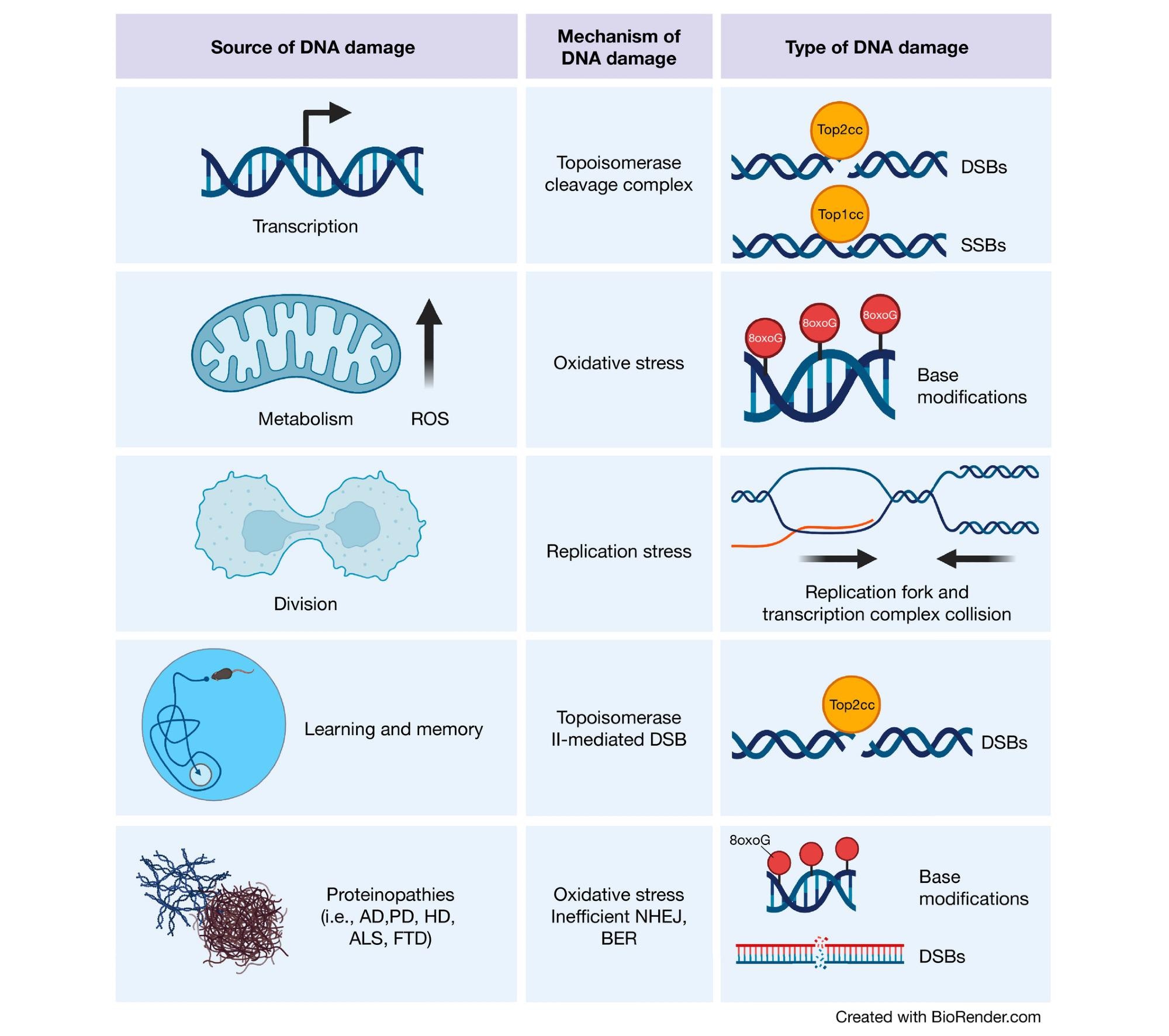
Sources of DNA damage in the brain Transcriptional activities can result in topoisomerase cleavage complexes, which lead to the induction of SSBs or DSBs depending upon the topoisomerase in question. Additionally, metabolic activity by mitochondria generate ROS, which can scar DNA bases with oxidative modifications. Although less common in the adult brain, cell division is also a source of DNA damage. Proliferation increases the chance of replication fork and transcription complex collision, thereby inducing DSBs. In the developing brain, this is a particular risk for NPCs, which harbor increased translocations in long genes (where these collisions are most likely to occur) important for neuronal function. Cognitively demanding tasks recruit specific neuronal ensembles whose plasticity is highly dependent upon immediate early gene transcription. Therefore, neurons generate topoisomerase II-mediated DSBs in response to learning and memory. Finally, the proteins responsible for various neurodegenerative diseases have also been found to play roles in DNA damage detection and repair. (Created with BioRender.com).
Single-Strand Breaks
Most genomic DNA injury is mediated by reactive oxygen species (ROS), which causes single-strand breaks (SSBs). This pathway occurs mostly in the nervous system because of the high need for mitochondrial respiration within this system, accounting for a fifth of the total oxygen uptake by the body. Mitochondrial metabolism may give rise to metabolic activity.
Both direct and indirect mechanisms exist for ROS-induced SSBs. With the latter, the strand is broken as a result of the need to repair a toxic DNA modification, such as the very common 8-oxo-7,8-dihydroguanine (8oxoG) that disturbs gene transcriptional processes and can induce mutations if wrongly repaired, accelerating brain aging and disease. This is part of a category of modifications called non-bulky base modifications, repaired by base excision repair (BER), including excision, and either short- or long-patch SSB repair (sp- or lp-SSBR, respectively).
Ultraviolet-induced DNA damage is in the form of bulky lesions that distort the DNA helix sterically. This initiates detection and nucleotide excision repair (NER) by one of two pathways. A third method of direct SSB is by aborted topoisomerase I (TOP1) activation, leading to the formation of TOP1 DNA cleavage complexes (Top1cc).
In the presence of oxidative DNA damage, Top1cc fails to resolve, which means this is a particularly toxic form of neuronal injury. A genetic condition called spinocerebellar ataxia with axonal neuropathy (SCAN1) is due to the absence of the SSB repair enzyme that resolves this injury.
Double-Strand Breaks
Double-strand breaks (DSBs) are likely to be more toxic though less common than SSBs, though they play a physiological role in, for instance, generating the vast spectrum of diversity of T cell receptors and antibodies, as well as in meiotic recombination. In actively replicating cells, DSBs are more common, probably because the replication fork collides with the transcriptional complex.
However, most DSBs occur because of transcriptional processes as a result of topoisomerase cleavage complexes. DSBs can arise due to topoisomerase II (TOP2) cleavage complexes intended to relieve torsion and expose certain genes for transcription due to the deficit of the Tyrosyl-DNA Phosphodiesterase 2 (TDP2) enzyme that should immediately resolve these complexes.
DSB repair occurs via on-homologous end joining (NHEJ) and homologous recombination (HR). The second method uses sister chromatids as the template for the synthesis of DNA to repair the breach, and is thus error-free but can occur only in replicating cells or after the S phase. The major DSB repair pathway is thus NHEJ for most post-mitotic cells. Single-strand annealing (SSA) is an alternative pathway, but like NHEJ, is error-prone in contrast to HR.
Functional Breaks May Cause Dysfunction Later
DNA breaks may have a physiological role in neuronal activity, but when they occur in neuron regulatory sequences, they could induce mutations and translocations, causing perturbations of transcriptional pathways that affect synaptic signaling. Several DNA repair mapping technologies are being developed or are already in use, and confirm this hypothesis. Poly(ADP-ribose) polymerase 1 (PARP1) and Ataxia telangectasia mutated (ATM) are two major sensors of SSBs and DSBs.
Neurological Diseases Due to DNA Repair Syndromes
Many neurological diseases are now known to be due to heritable DNA damage disorders, causing age-related or neurodevelopmental sequelae. These include SSBR protein mutations that cause loss of function and manifest purely as neurodegenerative disorders, including Ataxia with Oculomotor Apraxia types 1 and 4 (AOA1, AOA4).
SSBs can also form DSBs, which, in neurons, cannot be repaired by HR. Defective SSBR can trigger neurotoxicity via several mechanisms. For one, without the repair protein XRCC1, PARP1 becomes hyperactive, causing the accumulation of poly(ADP-ribose). This in turn depletes NAD+/ATP in the cell and causes cell death via the Parthanatos pathway.
Several mechanisms of PARP1-induced neurotoxicity have been elucidated, including dysregulation of DNA repair, suppression of transcription, and the accumulation of SSBs at enhancer sites that regulate neuronal activity.
DSBR mutations can also cause neurological syndromes such as ataxia-telangiectasia due to mutations in ATM kinase. Multiple mutations may cause a lower threshold for neuronal genomic instability causing deficits of neurological structure and function, unlike mutations in ATM genes alone. This could be because the cerebellar Purkinje cells have open stretches of chromatin that are sensitive to aberrations in DNA break repair mechanisms of certain kinds, causing deficient expression of Purkinje cell genes and the characteristic cerebellar atrophy and ataxia.
Neurodegenerative Disease and DNA Damage
Oxidative damage is seen in aging, and in brain tissue of patients with neurodegeneration such as in Parkinson’s disease (PD) or Alzheimer’s disease (AD). This is related to the increase in 8oxoG with age, which is mitigated by the histone deacetylase HDAC1. Single nucleotide variations (SNVs) can occur as a result of high 8oxoG levels, which perturb gene transcription at many levels.
This includes suppression of transcription factor binding, aberrant BER, and methylation of gene promoters. The resulting build-up of SNVs could cause genomic diversity in post-mitotic neurons, leading to neuronal dysfunction in the above conditions. In Huntington’s disease (HD) the hallmark age-dependent CAG expansion leads to toxic changes in the corpus striatum, due to the detection of oxidized or mismatched bases in the repeats by DNA repair proteins that then trigger unnecessary repair. Single nucleotide polymorphisms (SNPs) in DNA maintenance genes can lower the age of onset of HD, thus affecting its severity.
Importantly, in all these conditions, both SSBs and DSBs are found at high levels, and accompany significant markers such as cognitive impairment, in the course of the disease. This suggests that DNA strand breaks may contribute to disease progression. Many genes that are biomarkers of neurodegenerative disease caused by abnormal protein generation are involved in DDR pathways.
These and similar findings “have firmly established DNA breaks as a mechanism of neurodegenerative disease.” Further, genome-wide investigation of DNA breaks in post-mitotic neurons shows that the location of such breaks at regulatory regions that affect synaptic signaling and neuronal function is a major contributor to their toxic effects on the nervous system. That is, despite their functional relevance, such breaks may if improperly repaired, cause genomic translocation via erroneous translocations and mutations.
Neuroinflammation
Neuroinflammation is another key feature of neurodegenerative disease and is a major mediator of neurotoxicity. Many risk genes of AD actually affect microglial function, and these are brain-resident macrophages, associated with neuroinflammation. Senescence is possibly a driver of such inflammation and of neurodegeneration, and similar signals may emanate from neurons with DNA damage.
Such signals may arise through a variety of sources, including the activation of inflammatory signaling by senescence and DNA damage (via DDR pathways), and type-I interferon signaling because of cytosolic DNA fragments detected by the nucleic acid sensor cyclic GMP-AMP (cGAMP) synthase (cGAS) that activates Stimulator of IFN Genes (STING). This sets off multiple cascades of inflammation.
Cytosolic DNA also activates TLR9 (Toll-like receptor 9) or the NLRP3 inflammasome, all of which lead to neuroinflammation.
“It is clear that DNA damage can elicit neuron dysfunction broadly through two distinct mechanisms. First, the location of the lesion has a significant impact on transcriptional mechanisms required for normal cell function. Second, the downstream signaling pathways of lesion detection, whether they be through DDR or cytosolic nucleic acid sensing, can elicit apoptotic or inflammatory signaling that lead to neurotoxicity.”
-590_18da4e909d704b5fa71b7dd9ad43e610-80x70.jpg) Study reveals how DNA misfolding causes congenital heart disease
Study reveals how DNA misfolding causes congenital heart disease