Scientists have studied various aspects of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), the causal agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic, and developed pharmaceutical and non-pharmaceutical measures to combat the disease. They have characterized the pandemic by studying the repeated waves of COVID-19 cases due to the emergence of new SARS-CoV-2 variants. These new lineages exhibit greater viral fitness, where fitness is related to traits associated with lineage’s growth, including basic reproduction number (R0), the ability to evade immune responses, and generation time.
Background
As the SARS-CoV-2 lineages emerge, scientists warn that it is essential to identify them and predict the possibility of an outbreak. One of the main difficulties associated with this study is analyzing an ever-increasing, huge dataset, i.e., approximately 7.5 million virus genomes, constituting geographic and temporal variability.
Researchers stated that current phylogenetic approaches to assess the relative fitness of newly emerged lineages are computationally incompetent for evaluating large datasets, i.e., over 5000 samples. Furthermore, although ad hoc methods used to assess the relative fitness of specific SARS-CoV-2 lineages are computationally efficient, they depend on models that can compare only one or two lineages of interest to others. Therefore, it does not capture the dynamic complexity of multiple SARS-CoV-2 lineages circulating.
A mutation-based analysis could help identify specific genetic determinants associated with enhanced transmission and pathogenesis. This analysis would positively help predict the phenotypes of newly emerged lineages. For instance, D614G mutation in the spike protein of SARS-CoV-2 is associated with a high viral load. Other mutations found in the spike protein of the SARS-CoV-2 variants of concern (VOC), relative to the original strain, such as N439R, E484K, and N501Y, are linked to enhanced transmissibility, antibody escape, and increased affinity of the virus to bind to ACE2 of the host.
A New Study
Researchers believe that it is essential to identify functionally important mutations with the phenotypic outcome related to a large number of SARS-CoV-2 variants that have emerged since the beginning of the pandemic. Recently, scientists have modeled the relative fitness of SARS-CoV-2 lineages based on viral growth as a linear combination of the effect of each mutation.
In this new study in the journal Science, researchers have developed a hierarchical Bayesian regression model, named, PyR0, which can analyze the complete set of publicly available SARS-CoV-2 genomes. The authors stated that this model is also applicable to any viral genomic dataset. One of the advantages of this regression model is that it can estimate the growth rate of genomic sequences and, thereby, determine statistical strength among genetically alike lineages without entirely depending on the phylogeny.
The authors modeled the multinomial proportion of different lineages instead of an absolute number of samples for each lineage. In this study, they fitted PyR0 to 6,466,300 SARS-CoV-2 genomes, obtained from GISAID. The model contained 3,000 clusters derived from 1,544 PANGO lineages and 2,904 nonsynonymous mutations. The output of this regression model is a posterior distribution of the relative fitness of each SARS-CoV-2 lineage and the impact of each mutation on the fitness.
The computational challenge inherent in this large model led the researchers to use an approximate inference method, stochastic variational inference. This model helped predict the fitness of completely new lineages, infer lineage fitness, and estimate the impact of individual mutations on fitness.
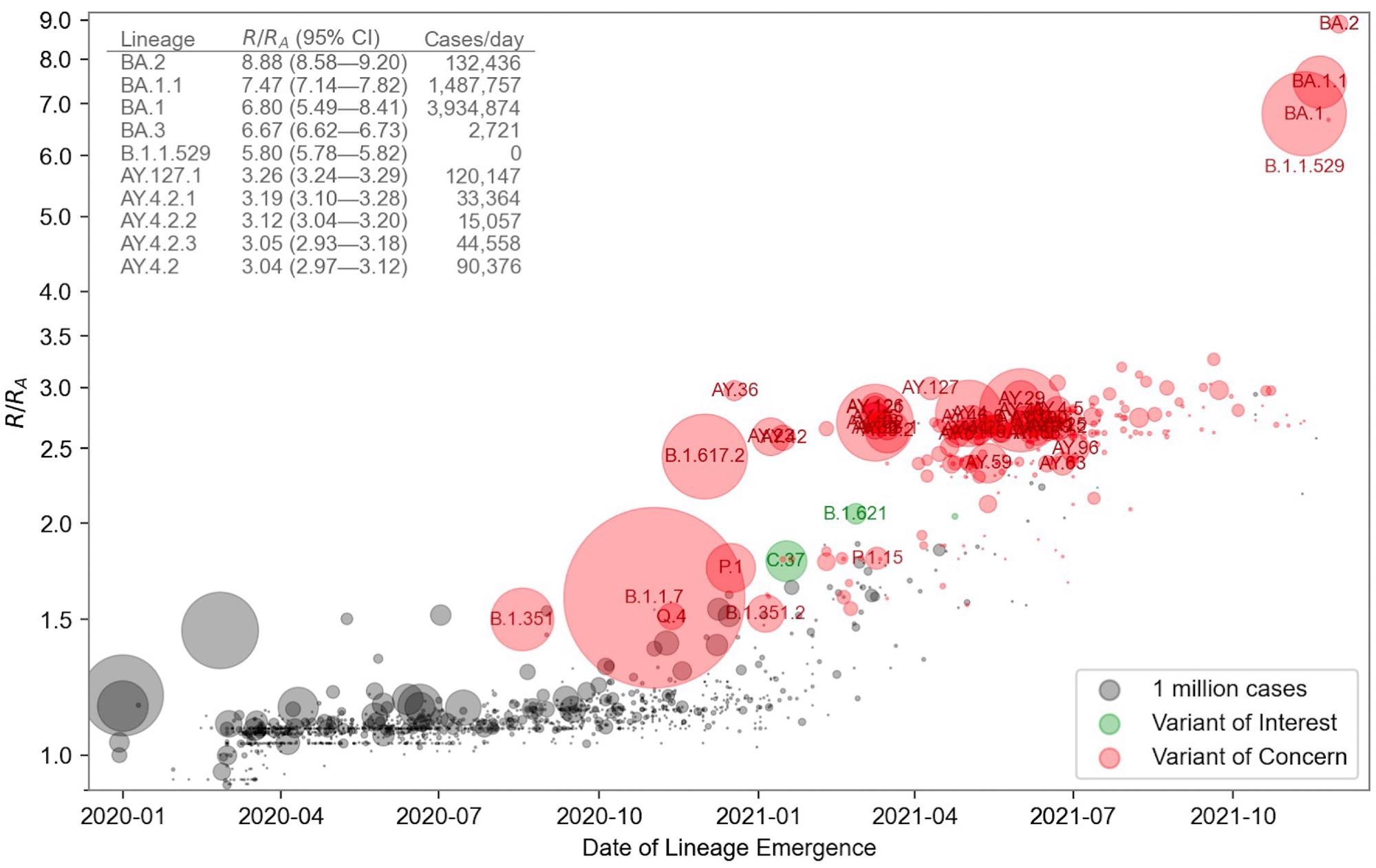
Relative fitness versus date of lineage emergence. Circle size is proportional to cumulative case count inferred from lineage proportion estimates and confirmed case counts. Inset table lists the 10 fittest lineages inferred by the model. R/RA is the fold increase in relative fitness over the Wuhan (A) lineage, assuming a fixed generation time of 5.5 days.
Key Findings
The model exhibited a modest upward trend over time among all lineages. Some lineages showed higher fitness over others. The qualitative uniformity of fitness estimates across spatial data subsets was determined via sensitivity analysis.
The rapid SARS-CoV-2 transmission in the human population from the beginning of the pandemic to early 2022 was marked by the rapid evolution of fitness and a surge in COVID-19 cases. Additionally, in some geographic regions, certain PANGO lineages with multiple successive peaks were observed, which suggested that lineages within them had varied fitness. This is the reason why researchers have algorithmically refined the PANGO lineages into finer clusters.
Importantly, scientists tested the predictive ability of the model and found that its predictions were reliable for one-two months into the future for SARS-CoV-2 VOC; predictions might differ for other new strains. The findings were consistent with the report of the World Health Organization that Omicron (PANGO BA.2) possesses the highest fitness among other VOCs.
In this study, PyR0 identified three hotspots in the S region that are associated with viral fitness. These regions are the receptor-binding domain (RBD), the N-terminal domain, and the furin-cleavage site. Researchers identified two mutations, namely, T478K and S477N, that significantly affect viral fitness. Additionally, PyR0 predicted the growth of a new SARS-CoV-2 variant.
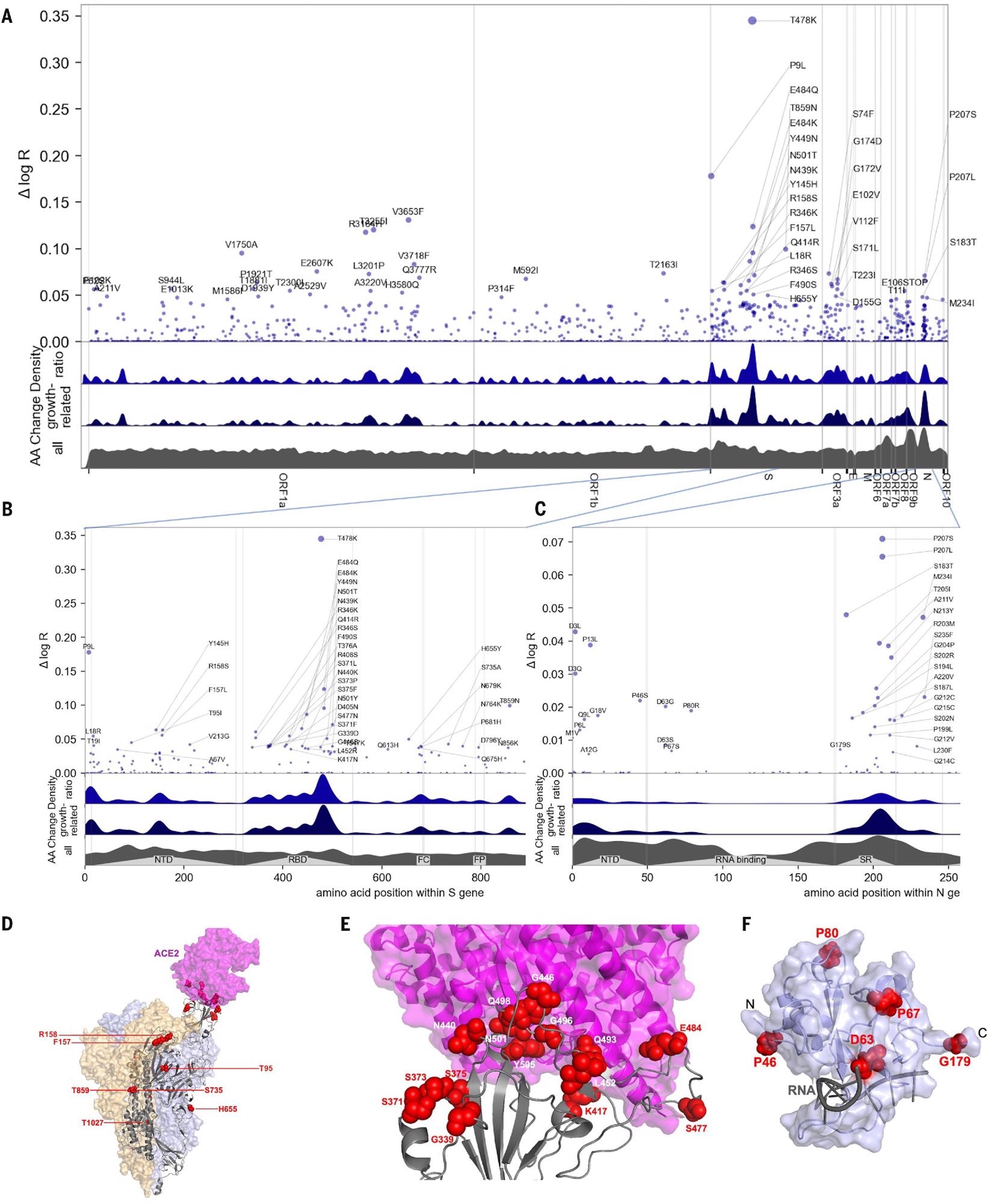 Manhattan plot of amino acid changes assessed in this study. (A) Changes across the entire genome. (B) Changes in the first 850 amino acids of S. In each of (A) to (C) the y axis shows effect size Δ log R, the estimated change in log relative fitness due to each amino acid change. The bottom three axes show the background density of all observed amino acid changes, the density of those associated with growth (weighted by |Δ log R|), and the ratio of the two. The top 55 amino acid changes are labeled. See fig. S13 for detailed views of S, N, ORF1a, and ORF1b. C. Changes in the first 250 amino acids of N. (D) Structure of the spike-ACE2 complex (PDB: 7KNB). Spike subunits colored light blue, light orange, and gray. Top-ranked mutations are shown as red spheres. ACE2 is shown in magenta. (E) Close-up view of the RBD interface. (F) Top-ranked mutations in the N-terminal RNA-binding domain of N. Residues 44-180 of N (PDB: 7ACT) are shown in light blue. Amino acid positions corresponding to top mutations in this region are shown as red spheres. A 10-nt bound RNA is shown in gray.
Manhattan plot of amino acid changes assessed in this study. (A) Changes across the entire genome. (B) Changes in the first 850 amino acids of S. In each of (A) to (C) the y axis shows effect size Δ log R, the estimated change in log relative fitness due to each amino acid change. The bottom three axes show the background density of all observed amino acid changes, the density of those associated with growth (weighted by |Δ log R|), and the ratio of the two. The top 55 amino acid changes are labeled. See fig. S13 for detailed views of S, N, ORF1a, and ORF1b. C. Changes in the first 250 amino acids of N. (D) Structure of the spike-ACE2 complex (PDB: 7KNB). Spike subunits colored light blue, light orange, and gray. Top-ranked mutations are shown as red spheres. ACE2 is shown in magenta. (E) Close-up view of the RBD interface. (F) Top-ranked mutations in the N-terminal RNA-binding domain of N. Residues 44-180 of N (PDB: 7ACT) are shown in light blue. Amino acid positions corresponding to top mutations in this region are shown as red spheres. A 10-nt bound RNA is shown in gray.
Conclusion
Modeling millions of viral sequences across multiple regions, PyR0 provided mechanistic insights into how mutations enhance viral fitness. Additionally, this regression model offered a panoramic view of viral evolution. According to the authors, their model could have accurately predicted or provided early warning regarding the detection of VOCs. As the model can be adapted faster to incorporate mutations of new linages, it can help protect the public from uncertainties caused by newly emerging variants.
 CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater
CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater