The SARS-CoV-2's NSP3 possesses a conserved Mac enzyme called Mac1. It is essential for the lethality and pathogenesis of the virus. Mac1 is a target that has emerged from SARS-CoV-2 that is both very challenging and appealing.
Despite having significant therapeutic potential, few small-molecule Mac1 inhibitors have been reported. Specifically, although Mac1 demonstrated a vital function in the pathogenesis of SARS in animal studies, no inhibitors of any form or reliable chemicals exist for the enzyme.
Fortunately, Mac1 quickly crystallized and exhibited high-resolution diffraction, enabling the fragment-based study of its recognition determinants computationally and empirically. Using this approach, the study's authors previously determined approximately 230 fragment structures. The ligands' binding positions tiled the enzyme's active site, yet despite frequently good ligand efficiencies, no fragments displayed affinities more robust than 180 μM.
About the study
In the current study, the scientists constructed the molecular determinants of Mac1, which were disclosed by the fragment structures, to find more potent compounds, moving closer to the development of chemical probes and therapeutic leads.
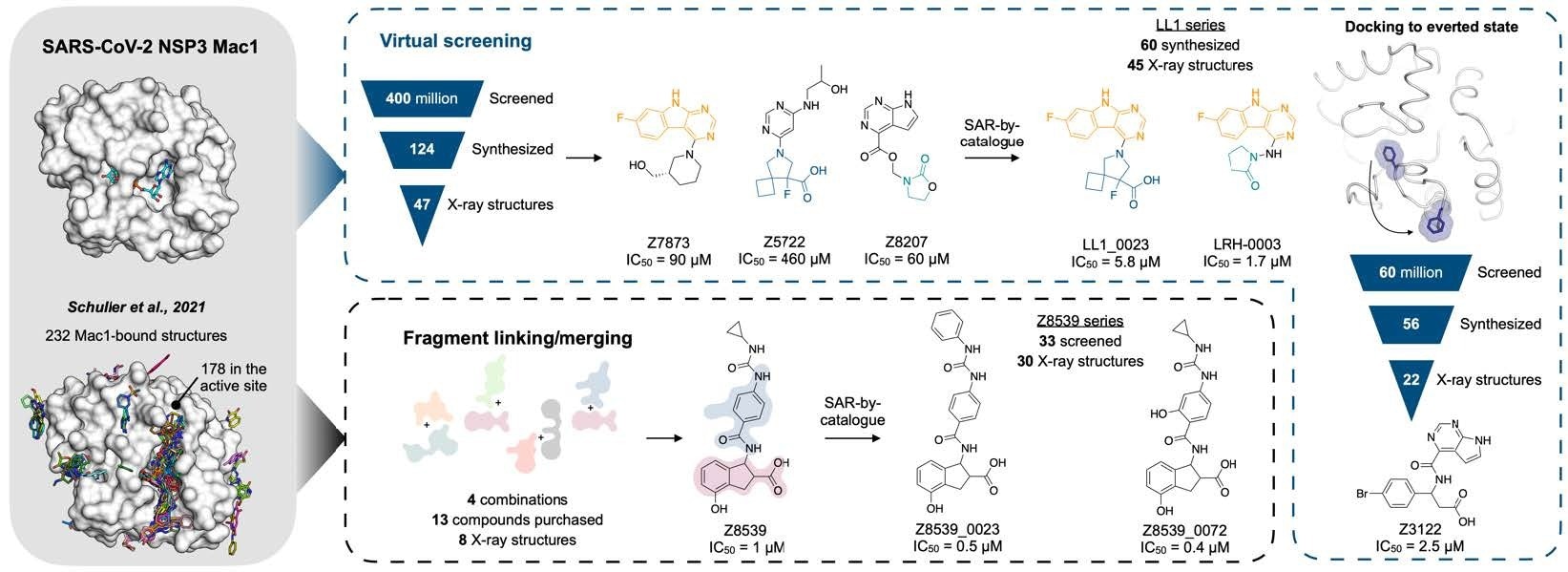 Overview of the structure-based strategies used to discover ligands that bind to the NSP3 macrodomain of SARS-CoV-2 (Mac1).
Overview of the structure-based strategies used to discover ligands that bind to the NSP3 macrodomain of SARS-CoV-2 (Mac1).
The team describes the structure-based creation of a range of chemical scaffolds possessing low-to-sub-micromolar affinities for Mac1 via iterations of computer-assisted design, structural profiling through ultra-high-resolution X-ray protein crystallography, and attachment assessment with in-solution tests. They established effective scaffolds using ultra-large library docking panels with over 450 million compounds and in silico linking of previously acquired fragment hits.
Overall, the present work reports the efforts employed for optimizing priorly defined molecular Mac1 determinants via two methodologies. Initially, the team attempted to link and combine pairs of fragments to produce large compounds that took advantage of many hot spots, reaching high affinities. For this, they applied a novel fragment-linking approach, exploring a virtual collection of 22 billion easily synthesizable molecules.
In the second strategy, the scientists used the hot spots identified by the first fragments to direct the computational coupling of extremely extensive chemical pools of lead-like compounds, which may have greater potency than the fragments coupled in their initial investigation.
Results and conclusions
The authors reported that 160 Mac1 inhibitory hits consisting of 119 unique scaffolds were found in the present investigation. Furthermore, the crystal structures of 152 Mac1-ligand complexes were identified, often with a resolution of 1 Å or higher.
The team discovered multiple ligands that caused a significant re-arrangement of the Mac1 active site loop, comprising residues 127 to 136. Relative to the 7 to 12 Å shifts in Phe132 shown in the study, conformational alterations involving Phe132, Ala129, and Asn99 were identified in this loop in the adenosine diphosphate ribose (ADPr)-attached condition and at low pH in the ligand-free enzyme.
Other macrodomains, such as human MacroD1 and PARP14, have also demonstrated everted loop conformations. Despite employing the ADPr-bound condition as a template for docking, the first virtual screening effort failed to find any molecules that stabilized the flipped conformation of Ala129 regardless of the apparent plasticity of this area.
Nevertheless, numerous structures having Ala129 in the flipped condition were discovered during compound optimization. These consisted of LL114 0041, which introduces a carboxylic acid into the phosphate-attaching subsite, and LL123 0020, stabilizing a water molecule in an identical position. Although the alteration in Ala129 was less significant, a comparable water network re-arrangement was observed for Z0828, the docking hit. These ligands present fresh possibilities for structure-informed design projects aiming at the Mac1's phosphate-attaching subsite.
The authors noted that the current attempt yields three key insights. Initially, molecules that incorporated important groups of pairs of fragments and were easily accessible via make-on-demand synthesis were found using an automated fragment-linking and -merging technique in conjunction with searches of ultra-large pools. These efforts resulted in the quick discovery of compounds with low μM affinity, later refined to much lower affinities such as 430 nM, e.g., compound Z8539_0072, a comprehensive improvement of >400-fold relative to the best initial fragment.
Second, templated again by the ligand-identification patterns disclosed by the fragments, molecular docking tests discovered molecules with affinities below 2.5 μM, with some in the mid-μM band, also optimizable to the low μM. Moreover, the best compounds exhibited ligand efficiencies superior to even the combined fragments.
Thirdly, despite most molecules being anionic harboring high polar surface areas decreasing cell permeability, structure-based optimization identified analogs like alcohols, ureas, and phenols possessing few hydrogen-donating groups, enabling swapping the anionic warheads for neutral ones. This shows that it might be possible to increase cell membrane permeability for several scaffold classes considered herein.
Altogether, the researchers stated that the structure-activity connections discovered through this work could serve as a model for future Mac1-targeting drug development. From a technical viewpoint, a dataset for benchmarking and enhancing computational tools for the discovery of drugs, such as free energy perturbation, was created by the abundance of structure-activity relationships paired with X-ray crystal structures for most molecules discovered in the research. The substances and configurations presented in this study will facilitate the development of first-in-class antiviral medicines that target the NSP3 Mac of SARS-CoV-2 from a therapeutic standpoint.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Structure-based inhibitor optimization for the Nsp3 Macrodomain of SARS-CoV-2; Stefan Gahbauer, Galen J Correy, Marion Schuller, Matteo P Ferla, Yagmur U Doruk, Moira Rachman, Taiasean Wu, Morgan Diolaiti, Siyi Wang, Jeffrey Neitz, Daren Fearon, Dmytro S. Radchenko, Yurii Moroz, John J Irwin, Adam R Renslo, Jenny C Taylor, Jason E Gestwicki, Frank von Delft, Alan Ashworth, Ivan Ahel, Brian K Shoichet, James S Fraser. bioRxiv preprint 2022, DOI: https://doi.org/10.1101/2022.06.27.497816, https://www.biorxiv.org/content/10.1101/2022.06.27.497816v1
- Peer reviewed and published scientific report.
Gahbauer, Stefan, Galen J. Correy, Marion Schuller, Matteo P. Ferla, Yagmur Umay Doruk, Moira Rachman, Taiasean Wu, et al. 2023. “Iterative Computational Design and Crystallographic Screening Identifies Potent Inhibitors Targeting the Nsp3 Macrodomain of SARS-CoV-2.” Proceedings of the National Academy of Sciences 120 (2). https://doi.org/10.1073/pnas.2212931120. https://www.pnas.org/doi/10.1073/pnas.2212931120.
 SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity
SARS-CoV-2 rarely reaches first-trimester placentas but still disrupts early pregnancy immunity