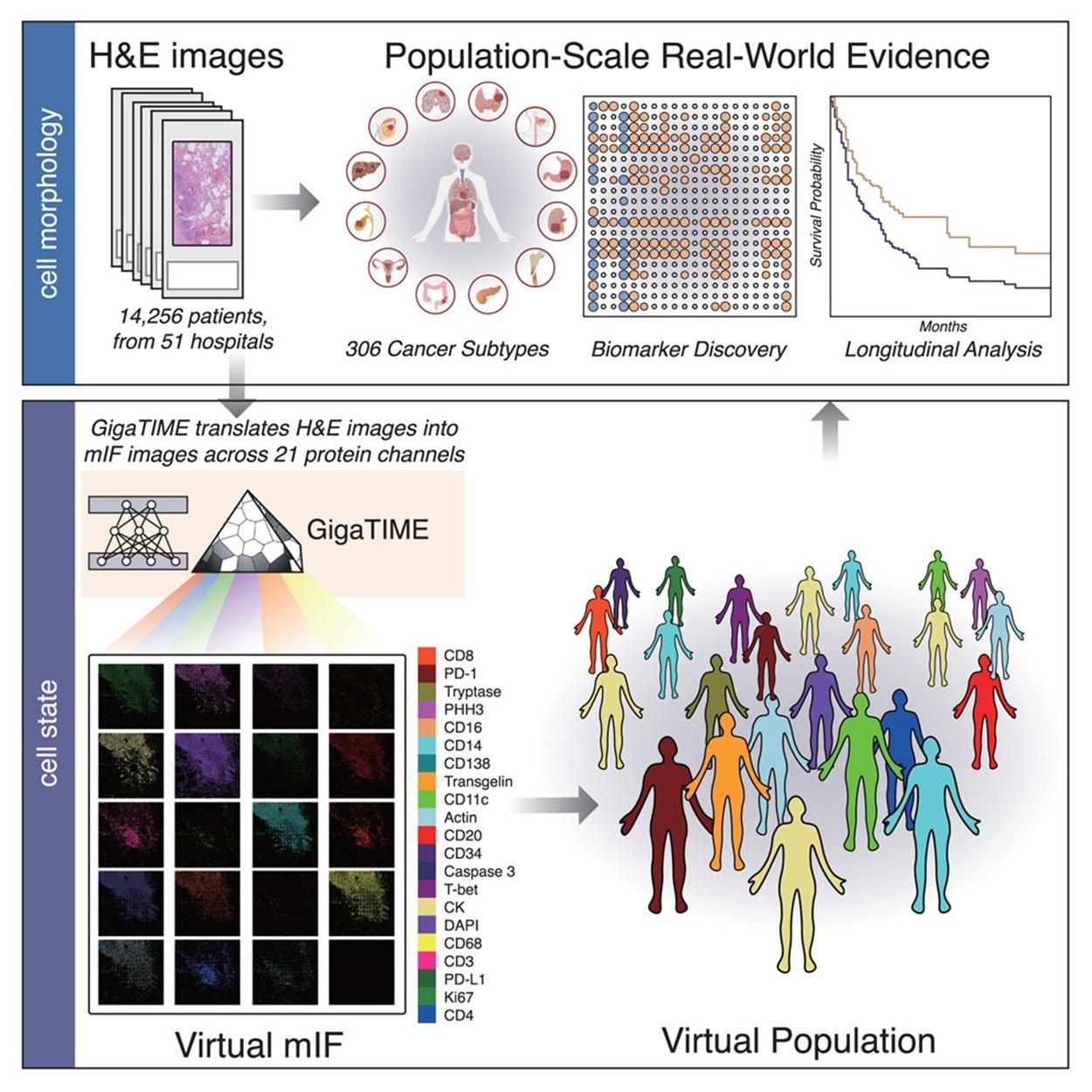
By transforming standard H&E slides into virtual multiplex protein maps, GigaTIME reveals how immune activity, tumor invasion, and survival are linked across thousands of cancers, opening a new path for scalable, data-driven oncology research.
Study: Multimodal AI generates virtual population for tumor microenvironment modeling
A recent study published in the journal Cell explored the capabilities of GigaTIME, a multimodal AI framework designed for large-scale modeling of the tumor immune microenvironment (TIME) in cancer research.
TIME Complexity and Profiling Challenges
The TIME is a highly complex spatial ecosystem composed of cancer cells and a variety of non-malignant cell types, such as cancer-associated fibroblasts (CAFs), immune cells, endothelial cells (ECs), pericytes, and others, all embedded within a remodeled extracellular matrix. It is profoundly associated with cancer progression, shaping tumor growth, invasion, metastasis, and therapeutic outcomes through its regulation of immune surveillance and facilitation of immune evasion.
Researchers employ immunohistochemistry (IHC) to characterize cell states within the TIME. For example, PD-L1 IHC staining is used to detect PD-L1 expression, a common biomarker for predicting response to checkpoint inhibitor therapies.
A major drawback of IHC is that protein activation is assessed individually, requiring a separate tissue sample for each analysis. This limitation poses a significant challenge for modeling the tumor microenvironment, as understanding the intricate interactions between tumor and immune cells requires evaluating multiple proteins simultaneously. Multiplex immunofluorescence (mIF) addresses this issue by enabling co-localized, multi-channel protein profiling on the same tissue section, while maintaining spatial organization.
Despite its promise, mIF is prohibitively expensive for large-scale studies, requiring costly reagents, specialized equipment, and labor-intensive workflows, thereby limiting dataset availability and clinical applications. In contrast, hematoxylin and eosin (H&E) staining is widely used and inexpensive in clinical practice to examine tissue and cell morphology. Although H&E images do not directly show cell states, their patterns may hint at them. AI models trained on many pathology images can detect features linked to where proteins are active in tissue.
AI-Based Virtual mIF Generation
GigaTIME generates diverse virtual mIF populations from large-scale H&E slides. Four hundred and forty-one mIF images were acquired from 21 H&E-stained slides spanning 21 protein channels to create the training dataset. After image registration and cell segmentation, this yielded a dataset of 40 million matched cells.
GigaTIME was applied to 14,256 whole-slide H&E images from Providence Health, which covered 24 cancer types and 306 subtypes across 51 hospitals and over 1,000 clinics. The model generated 299,376 virtual mIF images, resulting in a large multimodal dataset with associated clinical information. The analysis of the whole-slide H&E images produced a comprehensive TIME spectrum and atlas, and revealed more than 1,200 (specifically, 1,234) significant associations between clinical biomarkers and protein channels.
For each virtual mIF image, protein activation density scores were calculated and, subsequently, aggregated by cancer subtype to profile mIF-based TIME features. In addition to density, the authors quantified spatial metrics such as entropy, sharpness, and signal-to-noise ratio, which in some cases showed stronger associations with clinical biomarkers than density alone. GigaTIME was also applied to 10,200 TCGA tumors to generate 214,200 virtual mIF images, thereby demonstrating its robustness. The model's generalizability and reliability were confirmed by the strong concordance between datasets in protein activation patterns and biomarker associations.
GigaTIME’s performance in translating H&E images to mIF images was benchmarked against CycleGAN using three metrics at the pixel, cell, and slide levels. GigaTIME outperformed CycleGAN on 15 of 21 protein channels, underscoring the value of paired H&E and mIF data.
Out-of-sample analysis of GigaTIME’s generalizability was performed by testing it on breast and brain tumor microarrays that were not included in the training set. Despite diverse cancer types, stages, and sample formats, GigaTIME maintained strong performance in Dice scores and correlations, consistently outperforming CycleGAN and baseline methods across cancer types.
Stratified analysis by subcellular localization showed that nuclear proteins had higher translation quality than surface or cytoplasmic proteins, likely because their compact, well-defined structures are easier to predict. Some cytoplasmic and membrane proteins may be inherently less translatable from morphology alone, reflecting fundamental limits of H&E-to-protein inference.
Protein Signatures Linked to Tumor Invasion
GigaTIME identified spatial and combinatorial protein activation patterns and enabled risk-based patient stratification by stage and survival. Many associations differed by cancer type and histological subtype, highlighting the biological heterogeneity of the TIME. Validation using tumors from the Cancer Genome Atlas (TCGA) supported the generalizability of virtual protein activation patterns rather than directly validating staging predictions.
Studies conducted on GigaTIME virtual populations documented that, at the pan-cancer level, tumor invasion stage was associated with increased virtual PD-L1 activation and complex patterns of protein activation. This reflected a coordinated immune response. In advanced disease, the data suggested that alternative immune evasion mechanisms became increasingly influential over PD-L1-mediated pathways. The possible evasion of immune-induced apoptosis was indicated by reduced predicted cleaved caspase-3 expression.
Protein activations for multiple immune cell markers showed strong cross-correlation, supporting the need for immunotherapies targeting various cell types. The GigaTIME signature, combining all protein channels, outperformed single-channel models in predicting survival. Associations were found between GigaTIME virtual protein activations and both known and less well-described genomic alterations, as well as decreased immunogenicity linked to oncogene mutations such as KRAS.
For example, the authors highlight combinatorial relationships such as CD138 with CD68, and PD-L1 with cleaved caspase 3, illustrating how spatial and multiprotein signatures can reveal immune-tumor interactions not evident from single markers alone.
Expanding Access to Spatial Proteomics
Initial results demonstrated GigaTIME’s promise, with the largest virtual mIF association study to date. By enabling population-scale, spatially resolved proteomic inference from routine H&E slides, the approach has the potential to expand access to detailed tumor immune profiling in both research and clinical settings. However, there is a need for more geographic and ethnic diversity, as most patients were from the western United States.
The findings confirmed that H&E slides capture significant spatial proteomic signals, but translation quality varied across protein channels. This variability could stem from a number of factors, including varied tissue architecture, differences in underlying training data, biological heterogeneity, and marker-specific technical challenges. The authors note that certain proteins may be impossible to accurately infer from H&E morphology, underscoring the practical limits of virtual mIF.
Ongoing work aims to assess more protein channels, construct a comprehensive virtual mIF atlas, and incorporate cell segmentation models to shed more light on cell-to-cell interactions in the tumor microenvironment.
 New AI framework outperforms conventional biomarkers in predicting cancer survival
New AI framework outperforms conventional biomarkers in predicting cancer survival