A phage is a virus that infects a bacterium. People often get very confused about what the difference is between a virus and a bacterium. A virus, like a bacterium, is also a microorganism, but unlike bacteria, it needs to have a host to be able to replicate and propagate. All bacteria have these natural viruses, just like we have viruses that make us sick and our children sick.
All bacteria have their own sets of viruses, so they’re very specific. A virus, for example, that infects E. coli bacteria wouldn’t infect a different species of bacteria.
Over 90% of all viruses found that infect bacteria fall into three families or ‘types’ of viruses, but for the thousands of types of bacteria that exist, there are even more sub-types of viruses within these three families.
The three major families of viruses are known as myoviruses, siphoviruses, podoviruses. They’re double-stranded DNA viruses and have different shapes.
“Myo” means muscle and these viruses are have a long, contractile tail. Typically this type of virus is used to illustrate what a phage looks like. If you look up ‘bacteriophage’ on a search engine, you will see lots of images of myoviruses looking a bit like an ant or a lunar lander.
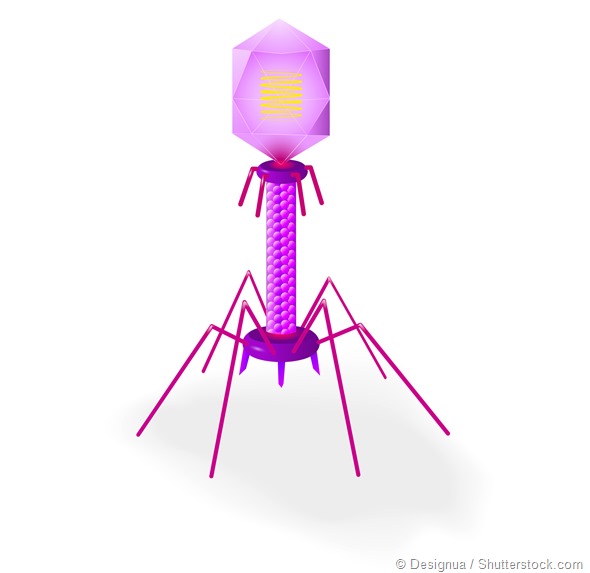
“Sipho” means siphon and siphoviruses have a long, non-contractile tails. Sometimes it’s flexible and sometimes it’s not. It is like a harpoon, in terms of the way it gets through the bacterial cell.
“Podo” means foot, and the podoviruses have got tiny little tails. In order to get into the bacteria, they often have enzymes associated with them that help them degrade the bacterial cell walls.
Within specific bacterial species, you’ll often find that some have got more of one family of viruses associated with them, but some bacteria are known to be infected with viruses of the three families.
How long have we known about bacteriophages and how has our understanding of them developed over time?
We have known about them for 100 years. They were discovered by an Englishman called Frederick Twort. He was working on smallpox vaccines which at the time could only be grown on cow hides and he was trying to work out why this was.
During this work he scraped cow hides to try to work out the ‘essential substance’ that made the vaccines grow and in the process he isolated Staphylococcus, which is often associated with hides.
Although the ‘essential substance’ is now known to not exist, he did notice “glassy” bits on the Staphylococcus bacteria that he realised were due to the destruction of bacteria. He also found that he could scrape these areas from one colony of Staphylococcus and use it to successfully infect another colony. He wrote up these observations in The Lancet, although but they were largely ignored.
Then a couple of years later, in 1917 bacteriophages were independently discovered by a French Canadian called Felix d’Herelle who was working in the Pasteur Institute. He was responsible for the name ‘bacteriophages’, which translates from Greek as ‘bacterial eaters’.
Quite quickly after finding these ‘invisible antagonistic microbes’ as he called the phages, he realized that they could have therapeutic uses, so he and colleagues developed bacteriophage treatments a couple of years after finding them.
The development of phages as therapeutics carried on for quite a long time, with people developing them in the Pasteur Institute and in many other places, during the ‘20s and ‘30s.
During this time a young scientist called Giorgi Eliava went to Paris and trained with a man called Felix d’Herelle. He learned lots about these viruses and then ultimately established what is now the Eliave Institute of bacteriophages, microbiology and virology in Georgia where he studied, developed and prepared bacteriophages to treat a wide range of topical, internal and respiratory bacterial pathogens.
However, with the discovery of antibiotics, research in phage therapy more or less stopped. There’s a great article that reviews this history . People carried on using viruses in Georgia and, in some other places such as France until about the 1970’s, you could buy many different preparations of viruses.
So although phages were used as a therapeutic in some places, in most places, there was a switch of focus from phage therapy research. People thought it wasn’t really necessary because we had antibiotics, which were seen as much more simple and easy therapeutics to use.
The emphasis in phage research between the ‘30s and ‘70s, focussed on using them as tools to discover the fundamentals of molecular biology. For example, our knowledge of DNA being genetic material, came from phage research.
In order to demonstrate this, Alfred Hershey and Martha Chase put phages in a blender and separated the proteins from the DNA in and showed that when they put DNA into the bacteria and you then got more viruses, but the same was not true for the proteins. Many other discoveries were also made such as the elucidation of messenger RNA.
In the ‘80s and ‘90s, things were taken even further because people started to look at novel enzymes that phages encoded. Lots of enzymes that are used in biotechnology and routinely in our labs all come from phages. For example, the enzymes that stick chunks of DNA together (ligases) when we perform cloning most commonly from viruses.
Then in the 1990s, which is when I was getting interested in viruses myself, there was suddenly a wider interest in phages. One driver for this, was the fact that scientists started sequencing bacterial genomes and they kept seeing a virus genomes within the bacterial DNA.
The first bacteria that they did this with was Haemophilus influenzae, and as more and more bacteria were sequenced, more and more viruses were found in them. People then began to wonder what all these viruses were doing within the bacteria?
Another reason for the renewed interest in phages came from ecologists who were interested in microbes in the wild. In doing this work they carried out estimates of viral abundance associated with bacteria. They were initially really surprised that for each bacteria that existed, there were between 10 and 100 viruses. This was shown in ocean environments, in rivers and in terrestrial environments and the numbers and ratios were roughly the same.
This question of what all these viruses were doing in the bacteria was a real driver in the ‘90s and over the following 15 years and scientists realized that they are the most abundant organism in the ocean where they played critical roles in shaping the nutrient cycling and evolutionary dynamics of the organisms they infect.
One example of the interlinked nature of phages in these systems can be seen from phages that infect marine cyanobacteria (blue green algae). These are really important photosynthesisers in the oceans and the viruses that infect them have actually acquired photosynthesis and other bacterial genes in order to maximise their own success.
Returning to now, I’d say what has motivated the current interest in phages is the awareness of antimicrobial resistance. Now, we can use our knowledge of phage ecology and all the things we’ve learned about from sequencing genomes and molecular biology, to direct us to where we are likely to identify new viruses and inform how we characterize them.
Please can you give an overview of your recent public lecture: 'The Enemy of My Enemy is My Friend: Developing viruses as novel therapeutics to treat bacterial infection’?
In that lecture, I tried to start off by setting the scene. I talked about the fact that we now realize that in the human body, we have 10 times more bacterial cells than human cells. That means we will have viruses in there doing interesting things, too.
I made the point that for a lot of the bacteria in our bodies, we’re becoming aware of the roles they’re playing, owing to projects such as the Human Microbiome Project.
The problem is that if we take an antibiotic, we wipe out these useful bacteria. There are two problems with antibiotics. One is the straightforward fact that they’re just becoming less effective because we’ve overused them and the second is that we’re starting to realize about the secondary effects antibiotics have.
Once you take a course of antibiotics, it takes a long time for the normal microbiota to re-establish. If bacteriophages were developed as therapeutics, they could not only be effective, but they wouldn’t be associated with these complications. They would quite specifically remove the pathogen and leave the other bacteria intact.
In the main body of my lecture, I outlined what I thought needed to be done to progress phage therapy. I tried to give a context and talk about where I saw the blockages in the exploitation of phages for therapeutic agents, and how the work in my lab is trying to address some of those issues.
To do this I introduced the audience to the concept of phages and showed how they work. I talked about the fact that they are still used in some places such as Georgia and Poland, but not in the Western Europe or more widely. I also talked about some of the reasons as to why there’s a real paucity of clinical trial data.
I briefly covered the history of phage therapeutic use and one example that I gave to illustrate how phages work was from a study where natural phages were used ‘accidently’ around 10 years ago in Bangladesh. This work is published in PNAS and details the work of a doctor who was trying to treat people with cholera. They were resistant to all the antibiotics he had access to, which meant he just saw more and more people coming in, infected with cholera.
Then he found that they all started to get better and he realized that the reason was because they’d all been drinking the same dirty water that gave them the cholera in the first place.
The numbers of bacteriophage had built up in that dirty water. He drew a correlation between the amount of phages in the water and the people who started to get better. They also had these viruses in their stools. It’s a really nice example of how phages are working naturally in disease .
I then talked about what we would need to do to get to a point where we’re not just letting nature take its course, but where we’re actually positively impacting, with sets of phages that could be used in this sort of way. The blockages are in part due to the fact that it can be quite difficult to identify bacteriophages and to just have good sets of phages that will target your bacteria.
Unlike a set of antibiotics, where you don’t particularly need to know what the bacteria is and just simply need to know that the antibiotic works, a tighter understanding of a pathogen is required with phages. If you want to treat someone with E. coli, it won’t work if you give them phages for Pseudomonas, for example. You need good, well-characterized set of bacteriophages.
You then need to have data on how best to deploy them to a diseased human. You need to know how many phages to add, what dosing regime, and how you give them in a stable formulation.
I talked about how I recently found viruses for the diseases I’m interested in. Since I’ve done a lot of ecology and environmental microbiology, I talked about how this fed into being able to find the right types of viruses. For example, if you go to an environment that’s really rich in a bacterial species, you’re more likely to find a good set of associated viruses and you can then pull them out and purify them.
Then I talked about how you actually determine the potential efficacy of bacteriophages. There’s no point in just showing how a virus works in a little flask that you’ve got your bacteria in when bacteria are all growing well. Under these conditions you quite possibly would kill all the bacteria. However, in order to test their efficacy under conditions where we are likely to want to use them, we need to try and mimic a system that’s similar to how the infection is found.
For example, we need to determine how well phages work in biofilms (how infections often occur) or on cell lines or other models. For example, we’ve got nice cell line models where we can grow gut epithelial cells that we then grow the bacterial pathogens on, and see how the phages work there. We also have insect models. I talked about how we use all these different types of models and the types of data we get. I showed that both biofilm, cell-line and insect models are all useful and that have used them to demonstrate phage efficacy.
I finished my talk by giving some examples of animal models that we’ve been using for our Clostridium difficile (C.diff) work, which is the phage program I’ve taken the furthest. I talked about some of the data that we’ve generated and how we are now working with a commercial company called AmpliPhi to develop a set of phages therapeutically. We have shown we have a suitable set of clinically relevant and prevalent phages .
I also presented some of the data on efficacy and how in a more complex model, we’ve shown that we can reduce carriage within C. difficile, this work was done with the help of collaborators from the University of Glasgow, led by Dr Gill Douce.
Finally, but importantly I talked about some work with Pseudomonas aeruginosa, which is a serious respiratory pathogen. I’m working with Prof Chris O’Callaghan from University College London/ Great Ormond Street Hospital, and we’ve got some nice data in this system. We’ve recently published an article that shows we can nebulize bacteriophages to the right place in the lungs and we’ve shown efficacy in a lung epithelial model.
How close are we to running out of antibiotics?
It’s different for different bacteria. There’s a lot of information on the World Health Organization website. They cite the main groups of bacteria where there’s a particular problem. It’s estimated that 18 strains of bacteria have now got complete drug resistance.
There’s a particular group that people talk about called ESKAPE (Enterococci, Staphylococci (MRSA), Klebsiella, Acinetobacter, Pseudomonas, Enterobacteriacae). There are a number of groups of bacteria that are a concern, where we have already more or less got to the end of the line.
There are some quite shocking statistics. It’s thought that half of the people that die from a bacterial infection in Europe each year have got multi-drug resistant bacteria. It is a major problem.
Why are bacteriophages more complicated to use than standard chemical antibiotics?
A standard antibiotic is a chemical that can be applied to a bacterial cell where it will target one aspect of bacterial growth or replication that a bacteria needs; whereas a bacteriophage is a replicating, evolving organism that needs to attach to, and insert itself into a bacteria and then kill it. So there are a relatively complicated set of process to unravel that are involved in bacteriophage-based killing.
Specificity is also an issue. You need to know if the bacteria you want to treat is sensitive to that virus whereas antibiotics often have a wider target specificity. This isn’t necessarily a disadvantage, but it does add to the complexity of treatment and highlight the need for an appropriate diagnosis to be made.
Then there’s fact that a bacteriophage-based treatment is always evolving; it will evolve with your bacterial host. Although phages may might kill the bacteria initially, just like antibiotics, once they are used the bacteria will start to develop resistance against it and will then start evolving. The phage will then to counter this and phages will evolve to target the resistant bacteria.
So essentially with phages you’ve got a dynamic entity, which is different from having a straightforward chemical. You need to understand the way in which your viruses will be interacting with your bacteria in order to do things like minimize the build-up of resistance.
One of the ways that scientists work out how to reduce resistance relates to your first question about how many types of bacteria there are. One thing that is generally agreed on within the phage research community is that if we can find different types of bacteriophages, and have a mixture of them, we can put the phage mixture into bacterial cells. They’ll then all be killing the bacteria in slightly different ways, which means that we’ll likely be able to reduce resistance.
We’ve seen that with our C. difficile data, and others have seen it in different systems. If we add different types of viruses to C. diff, we can completely eliminate it in vitro, but if we have only add one type of phage, the bacteria can evolve resistance.
I also think there are straightforward things that make bacteriophages difficult to use such as shelf-life, stability, and the making of a standard product.
There are benefits too though. Phages can penetrate biofilm. Often antibiotics don’t work in a biofilm because they can’t get through the polysaccharide and sugary DNA layer that’s extruded by the bacteria, whereas phages have the enzymes that can actually get through these biofilms.
How can genome information be used to inform bacteriophage therapeutic development?
I think this is going to be key. A hundred years ago when phages were first developed as therapeutics this information was not available. Scientists showed that the viruses killed bacteria but there was no mechanistic understanding.
Now we’re in an amazing situation where we can get the necessary information to work out how phages are killing their hosts and how the arms race between bacteria and phages is played out.
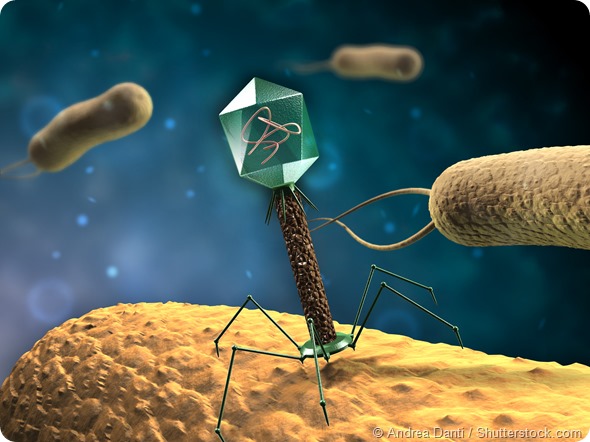
Around 15 years ago, I sequenced my first virus and it cost £26,000 and took 6 months. These days, it costs a couple of hundred pounds and so we can get the information we need much more quickly.
An important thing we do when sequencing the genomes is to make sure there’s nothing bad in there. Clearly, you don’t want to put the virus into a bacteria that encodes toxins or the various other genes that are not conducive to phage therapy. Also, you want to develop a , diverse set of viruses so you can use the genome information to ensure there is a genetic basis for their diversity.
It might be that you can only find myoviruses that infect pathogens, but there are different types of myoviruses. You can use that genome information to establish that they’re all quite different subclasses. You can also use the information to start to find out what new genes are present.
We can also use the genomic information for the quality control of a product. A hundred years ago, you’d produce your viruses and put them into vials to be used but it would have been hard to standardise a product. How would you know that that virus preparation you produced in the end is exactly the same as the viruses you seeded production with? The genome information is going to be really useful for the QC essentially of an end-product.
The first generation of bacteriophages that are being trialled at the moment, and will be in the near future, are viruses that occur naturally. They’ve been selected because they’re the aggressive winners of a natural bacteriophage war and then they’re purified and put into a situation.
The second-generation viruses will likely take advantage of our ability to manipulate viral DNA . There are different things you can do with natural viruses. You can engineer them, put different genes into them and for example to have a situation where you have fewer viruses with the same spectrum of activity. .
Ultimately, the genome information will feed into those second generation bacteriophages, the engineered viruses. We’ll be able to use the genomes to play with them and take bits out and see what happens when do this. Genome information will be brilliant for that.
What are the main hurdles to overcome in bacteriophage therapy?
I’d say there are two main hurdles. One is that for viruses to be used widely, we require funding for their development. Essentially, we need more clinical data because at the moment, the amount of research funding that’s gone into bacteriophages is tiny and we need more. We need clinical data to demonstrate efficacy in controlled environments.
The second hurdle relates to having an appropriate regulatory framework for phages to be regulated and licensed.
Regarding clinical trial and efficacy data, we’re now starting to see more small-scale animal trials and so on. I think that kind of work will then lead to more clinical trials. There are a few taking place this year that I know of.
There is a lot of data from places like Georgia where they’ve been researching for years and years. A lot of that work was translated. There’s a really informative Georgian book you can find on Amazon by Nina Chanishvili , that was funded partly by the British government to translate all of their early work into English. There’s a lot of interesting data there.
However a lot of the data describes case studies and the way in which the data is collected is not the way in which data would be collected in the West for a clinical trial. There is of course a lot of expertise in Georgia, and a lot of really useful stuff that has been done. I’m lucky enough to enjoy collaborations with Georgian phage researchers who have taught me a lot.
There has also not been an awful lot of attention paid to how to actually deliver the therapy and how we are going to get them to the lungs or to the stomach, for example. I think there needs to be a lot more work there and more interactions between pharmacists, and researchers with translational experience.
As I said then there is what is commonly described as the ‘regulatory hurdle’. I have previously had grants that were rejected outright the funding agencies were worried by the lack of a clear regulatory path to take a product to market.
Currently, under the European law, phages are regulated as a medical product. They are technically a ‘medical product’ or an ‘advanced medical product’, but there is an issue with both of those classifications,
s o there isn’t a particularly clear regulatory pathway at the moment, but a number of initiatives are engaging with European regulators and also, with the FDA. At the 21st Evergreen Phage meeting this year, FDA representative gave a clear and positive presentation on the FDA’s attitude to phage regulation.
Similarly at the ‘Phages in Interaction IV meeting in September this year, an EU representative spoke about about what could be done for better engagement in order to establish a regulatory pathway. It is not certain how phages will be regulated, but it does seem that there is a lot of engagement between regulators and people trying to develop them.
What do you think the future holds for bacteriophage therapy and antibiotics?
I think it’s a really exciting time to be studying phage therapy because we have the strong drivers of antimicrobial resistance that really focus our mind and encourage us to examine such promising antimicrobials. Similarly, initiatives to remove antibiotics from food production have motivated farmers and those in this industry to examine possible alternatives.
With this awareness has been an acknowledgement that funding is needed both from a medical perspective and a food and agriculture perspective. This has translated into funding being made available from research councils, government initiatives, charities and companies. You can see the amount of scientists doing this research is increasing because now we have the financial driver and the tools to be able to progress.
It’s also exciting because there are lots of things that we can actually do now and various different types of tools available so that we can sequence and look at RNA, proteins and the interactions. It’s a good time to be studying phage therapy and I think there’ll be a synergism between how phages are used in systems where perhaps the regulatory environment is not as strict, such as the agricultural and animal sectors, and those where it is a lot tighter such as phages for human medical conditions. When we see positive results in animal/agricultural systems, that’s going to add to the body of evidence of phage efficacy.
For example, you can see phages already being used in meat and cheese. The European food regulators allow Listeria phages to be used in cheese products and the FDA allows them in processed meats such as hot dogs. They have a status that is referred to as GRAS or Generally Recognized As Safe.
So although there are no phages being used clinically at the moment, they are being used in other ways. I think we, as a phage community, need to work out how it’s best to use them and what the best targets are for using phages, initially. Then, as efficacy is robustly demonstrated, scientists can develop phages for more complicated uses where they could be incredibly valuable but may need more development time.
It would of course be foolish to abandon antibiotics and researchers are currently trying to work out how we can preserve the antibiotics we’ve got, as well as developing new ones. Using phages, and using antibiotics, are not necessarily incompatible activities.
There probably will be some cases where the use combinations of antibiotics and phages will be optimal. We did some work with Pseudomonas where combining antibiotic treatments with phages was highly effective. You can imagine situations where someone’s got a chronic infection; you could use phages for those conditions and then keep antibiotics for the acute infection.
It will not be as simplistic as phages overtaking antibiotics, antibiotics have been and will continue to be invaluable in the treatment of bacterial infection, but as they lose efficacy, it is necessary to look for alternatives and I think that phages, and phage based products have a lot potential to be developed as therapeutics, and they can help us preserve, and can extend our antimicrobial armory.
Where can readers find more information?
For more information on antibiotic resistance:-
About Professor Clokie
I obtained a BSc in Biology from Dundee University in 1996 an MSc in Biodiversity from Edinburgh University in 1997, and a PhD from Leicester in Molecular Ecology in 2001. I then did 6 years of Post-Doctoral research at the University of Warwick and in Scripps, La Jolla, San Diego.
In 2007 I was appointed as a lecturer at Leicester, in 2011 a Reader (Associate Professor) and this year I was promoted to Professor in Microbiology.
My research focuses on phages that infect bacteria of medical and ecological relevance and I have published 41 papers in this area. I am interested in the ecology and evolution of viruses, and how and where they evolve, and in determining the mechanistic basis for the way in which they impact bacterial hosts. So to do this the work transcends from genome sequencing, through transcriptomics and proteomics.
I also like to develop relevant model systems in which to study host-phage interactions. I am particularly interested in phage therapy and how a knowledge of phage behaviour in the environment and in model systems can inform phage therapy development.
My major focus has been on Clostridium difficile where I have isolated a large phage collection. In vitro and in vivo data has shown that the viruses have therapeutic potential. I have filed a patent on these phages and am working with AmpliPhi to develop a product.
In my laboratory we also have projects on the characterization, biology and development of other bacteriophages including Pseudomonas aeruginosa, Salmonella and of phages that infect a range of other serious, yet neglected pathogens including Borrelia, Brachyspira, Burkholderia pseudomallei and Streptococcus pneumoniae.
I have had regular contact with BBC and other press to talk about my work and other phage projects, and have consulted to Science museum, London and Eden Project to advise on bacteriophage displays.
 Gut microbes emerge as potential players in estrogen-driven cancers
Gut microbes emerge as potential players in estrogen-driven cancers